






Pioderma gangrenoso (PG) é dermatose rara frequentemente associada a doenças sistêmicas. Observam‐se mecanismos autoinflamatórios com infiltrado neutrofílico e necrose. Em virtude da alta morbidade, resposta variável à terapia e falta de padronização do tratamento, o PG constitui condição altamente complexa.
ObjetivosAvaliar as características epidemiológicas, clínicas e laboratoriais, e a resposta à terapia de pacientes com PG acompanhados no Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo.
MétodosEste estudo retrospectivo e descritivo incluiu pacientes com PG confirmado em seguimento no Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo de janeiro de 2000 a agosto de 2021. Os dados foram obtidos de prontuários médicos.
ResultadosForam incluídos 50 pacientes neste estudo. O tempo médio de início dos sintomas até o diagnóstico foi de 26,5 meses. As lesões ocorreram predominantemente na extremidade inferior em 72% (n=36/50), e o tipo ulcerativo foi o mais comum (n=43/50; 86%). Dor local foi mencionada em 39/50 (78%), e 12/50 (24%) apresentaram patergia. As doenças associadas mais frequentes foram doença inflamatória intestinal (n=10/20; 20%) e hidradenite supurativa ([HS] n=10/20; 20%). Corticoide sistêmico em altas doses foi, em geral, a primeira terapia (88%), isoladamente (n=7/50; 14%) ou em associação com imunossupressores clássicos ou imunobiológicos (n=37; 74%). A maioria dos pacientes (n=32/50; 64%) teve pelo menos uma hospitalização. Controle da doença foi alcançado em 44/50 (88%) pacientes, com recorrência em 48% (n=24/50) e cura total sem medicação em 24% (n=12). Dezesseis pacientes (32%) foram tratados com pelo menos um agente imunobiológico além dos medicamentos clássicos.
Limitações do estudoDesenho retrospectivo, descritivo e número de pacientes.
ConclusõesHouve longo atraso no diagnóstico, associação com condições sistêmicas e cutâneas, e necessidade de terapia imunomoduladora ou imunossupressora prolongada (agentes clássicos e também agentes biológicos) para controle do PG.
Pioderma gangrenoso (PG) é dermatose incomum, caracterizada pelo acúmulo de neutrófilos na derme, hipoderme e, raramente, em órgãos internos.1 O PG afeta de três a dez pessoas por milhão por ano,1 especialmente mulheres adultas, mas pode ocorrer em qualquer idade.2 As lesões de PG são consideradas manifestação autoinflamatória resultante da desregulação da imunidade inata e da superprodução de mediadores inflamatórios (como interleucinas 1 e 17, fator de necrose tumoral alfa) que promovem infiltração neutrofílica tecidual.1,3
A apresentação clínica do PG é polimórfica e inclui as variantes ulcerativa (ou clássica), pustulosa, vegetante e bolhosa (ou atípica). A forma ulcerativa é a mais comum, evoluindo de pápulas ou pústulas para úlceras dolorosas com borda violácea bem definida e borda solapada. Afeta principalmente os membros inferiores e é frequentemente associada à doença inflamatória intestinal (DII), artropatias inflamatórias, gamopatia por IgA e neoplasias. A variedade pustulosa, também associada à DII, é caracterizada por pústulas no tronco e membros inferiores que regridem sem cicatrizes ou podem progredir para PG clássico. A forma vegetante apresenta‐se como placa violácea única de crescimento lento ou abscesso, geralmente no tronco, que cicatriza com padrão cribriforme. O PG bolhoso é caracterizado por bolhas de progressão rápida, geralmente na face e membros superiores, que evoluem para necrose. É frequentemente associado a doenças hematológicas, como leucemia mieloide, linfoma, gamopatia monoclonal e síndrome mielodisplásica. O PG crônico que afeta a mucosa oral e labial é chamado de pioestomatite vegetante. O PG também pode se desenvolver em outros órgãos, incluindo pulmões, rins, ossos e olhos.4
O PG também pode estar associado a infecções por HIV e hepatite C, lúpus eritematoso sistêmico, diabetes mellitus e psoríase.5,6 Também pode ser induzido por medicamentos como cocaína, propiltiouracil e antipsicóticos. Pode, ainda, ocorrer no contexto de síndromes autoinflamatórias, como PAPA (artrite piogênica, PG e acne), SAPHO (sinovite, acne, pustulose, hiperostose e osteíte) e PASH (PG, acne e HS). Traumas, incluindo procedimentos cirúrgicos, podem piorar ou desencadear lesões, um fenômeno chamado patergia. PG pós‐operatório foi documentado após cirurgia ortopédica, cardiotorácica, geral, plástica e ginecológica. O desafio no diagnóstico surge da sua falta de identificação, sua tendência a imitar feridas infectadas e exacerbação após desbridamento (procedimento padrão para tratar infecções de feridas) em decorrência da patergia.6–26Quando há suspeita de PG, uma avaliação abrangente é recomendada, incluindo biopsias cutâneas para exame histopatológico e culturas para descartar etiologias infecciosas, neoplásicas e autoimunes. A histopatologia do PG não é específica, mostrando infiltrado inflamatório misto, e a avaliação sistêmica inclui a investigação de doenças associadas frequentes.8–12A terapia varia de acordo com a gravidade do PG, sua extensão, doenças associadas e tolerância do paciente, com o objetivo de reduzir a atividade inflamatória, promover a cicatrização da ferida, controle da dor e tratamento de condições associadas. Além disso, traumas como desbridamento cirúrgico e enxerto devem ser cuidadosamente indicados, pois podem piorar ou desencadear novas lesões.1,10 Corticosteroides sistêmicos e ciclosporina são considerados terapia de primeira linha. Outros imunossupressores e agentes imunobiológicos são frequentemente necessários em virtude da refratariedade das lesões do PG.8,12,27
O PG prenuncia alto impacto na qualidade de vida dos pacientes, seja pelas lesões dolorosas e desfigurantes ou pelas comorbidades associadas.4 O atraso no diagnóstico pela raridade da doença e inexperiência dos profissionais de saúde, o ônus socioeconômico relacionado ao tempo gasto com cuidados médicos, efeitos adversos dos medicamentos e internações hospitalares contribuem ainda mais para o aumento da morbidade do PG.1,3,9
O objetivo deste estudo foi delinear o perfil clínico, epidemiológico, laboratorial e terapêutico dos pacientes com PG em seguimento no Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo (HCFMUSP) de janeiro de 2000 a agosto de 2021.
MétodosApós aprovação do Comitê de Ética (CAAE: 57067622.0.0000.0068), este estudo retrospectivo e descritivo incluiu todos os pacientes com PG em seguimento no Departamento de Dermatologia do HCFMUSP de janeiro de 2000 a agosto de 2021. O diagnóstico de PG foi baseado em características clínicas, achados histopatológicos compatíveis e exclusão de outros diagnósticos diferenciais.
Os prontuários clínicos dos pacientes foram analisados para obtenção dos seguintes dados: variáveis sociodemográficas, hipóteses diagnósticas anteriores, início dos sintomas, número e localização das lesões, subtipos clínicos, sintomas associados, comorbidades, exames laboratoriais e histopatológicos, medicamentos, hospitalizações, tempo para controle da doença e recorrências.
As variáveis categóricas foram expressas como frequências e porcentagens. Médias, desvio padrão, mediana e valores mínimo e máximo foram calculados para variáveis quantitativas. A associação entre variáveis categóricas foi avaliada pelo teste Qui‐quadrado de Pearson. O nível de significância adotado foi de 5%. As análises foram realizadas no software estatístico SPSS para Windows v. 25.
ResultadosDados sociodemográficosDe janeiro de 2000 a agosto de 2021, 50 pacientes foram diagnosticados com PG. Houve predominância do gênero feminino (n=29/50; 58%). A média de idade no início dos sintomas foi de 33,7 anos (3 a 65 anos), e 87,8% se declararam brancos. Outras características sociodemográficas são apresentadas na tabela 1.
Gênero, idade no início dos sintomas e cor da pele dos pacientes
| Aspectos | n=50 | n (%) | nfaltante | |
|---|---|---|---|---|
| Gênero | Feminino | 29 (58,0) | ||
| Masculino | 21 (42,0) | |||
| Idade no início dos sintomas (anos) | Médias (DP) | 33,7 (16,0) | ||
| Mediana (mín‐máx) | 30 (3‐65) | |||
| Cor da pele (autodeclarada) | Branca | 43 (87,8) | 1 | |
| Preta | 4 (8,2) | |||
| Parda | 2 (4,1) |
PG foi a única hipótese diagnóstica em 24 de 50 (48%) casos. A média de idade no momento do diagnóstico foi de 36,2 anos (5 a 65 anos), com duração média dos sintomas de 26,5 meses (0,25 a 480 meses).
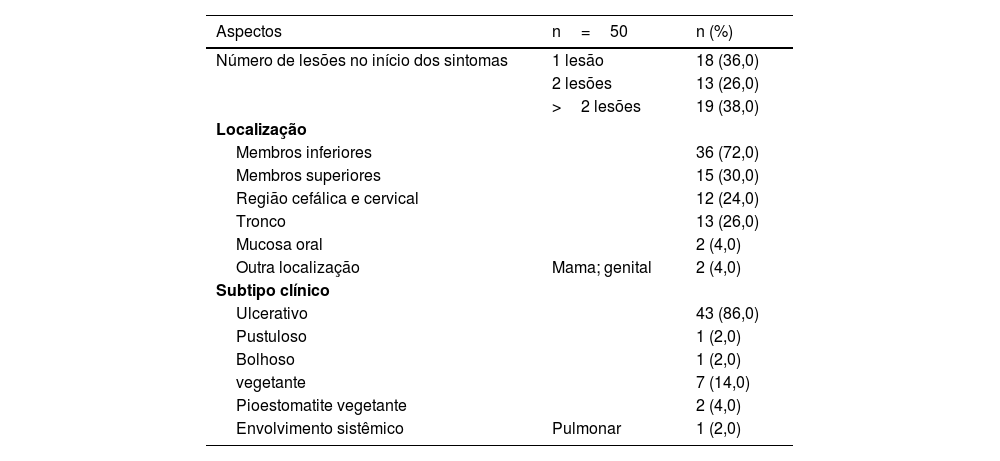
Lesões múltiplas (> 2) ocorreram em 19 de 50 (38%) pacientes. A região mais frequentemente afetada foi a extremidade inferior (n=36/50; 72%), seguida pela extremidade superior (n=15/50; 30%), tronco (n=13/50; 26%) e cabeça e pescoço (n=12/50; 24%). O subtipo ulcerativo (fig. 1) foi o mais comum (n=43/50; 86%; tabela 2). Dois pacientes apresentaram envolvimento oral e um teve envolvimento sistêmico (pulmões). Vinte pacientes (40%) apresentaram lesões em mais de uma localização; a associação de membros inferiores e superiores foi a mais frequente (n=7/20; 35%). Quando múltiplas lesões estavam presentes, o subtipo ulcerativo foi o mais encontrado (n=17/20; 85%). Os subtipos bolhoso e vegetante estão ilustrados nas figuras 2 e 3.

Número, localização e subtipo de lesões
| Aspectos | n=50 | n (%) |
|---|---|---|
| Número de lesões no início dos sintomas | 1 lesão | 18 (36,0) |
| 2 lesões | 13 (26,0) | |
| >2 lesões | 19 (38,0) | |
| Localização | ||
| Membros inferiores | 36 (72,0) | |
| Membros superiores | 15 (30,0) | |
| Região cefálica e cervical | 12 (24,0) | |
| Tronco | 13 (26,0) | |
| Mucosa oral | 2 (4,0) | |
| Outra localização | Mama; genital | 2 (4,0) |
| Subtipo clínico | ||
| Ulcerativo | 43 (86,0) | |
| Pustuloso | 1 (2,0) | |
| Bolhoso | 1 (2,0) | |
| vegetante | 7 (14,0) | |
| Pioestomatite vegetante | 2 (4,0) | |
| Envolvimento sistêmico | Pulmonar | 1 (2,0) |


Dor local, geralmente de extrema intensidade, estava presente em 39 casos (58%). Sintomas sistêmicos estavam presentes em 40 (80%) casos: febre em 12 (24%), dor nas articulações em 11 (22%) e perda ponderal em sete (14,9%) casos.
As comorbidades associadas são detalhadas na tabela 3.
Condições associadas
| Aspectos | n=50 (%) |
|---|---|
| Condições associadas. | 40 (80,0) |
| Doença inflamatória intestinal | 10 (20,0) |
| Doença de Crohn | 3 (6,0) |
| Colite ulcerativa | 7 (14,0) |
| Neoplasia de órgão sólido | 2 (4,0) |
| Próstata | 1 |
| Fígado | 1 |
| Doença hematológica | 5 (10,0) |
| Gamopatia monoclonal | 1 |
| Gamopatia policlonal | 1 |
| Mielofibrose | 1 |
| Síndrome mielodisplásica | 2 |
| Artrite | 6 (12,0) |
| Artrite lúpica | 1 |
| Artrite psoriásica | 1 |
| Artrite reumatoide | 3 |
| Artrite piogênica | 1 |
| Hidradenite supurativa | 10 (20,0) |
| PASH | 2 |
| PAPASH | 2 |
| PAPA | 1 |
| Doença autoinflamatória indefinida | 1 |
| Desencadeado por medicação | 2 (4,0) |
| Isotretinoína | 1 |
| Tocilizumabe | 1 |
| Doenças infeciosas | 3 (6,0) |
| Hepatite C | 2 |
| HIV | 1 |
| Outras dermatoses exceto HS | 7 (34,0) |
| Acne conglobata | 1 |
| Eritema anular centrífugo | 1 |
| Lúpus eritematoso | 1 |
| Psoríase | 1 |
| Pustulose subcórnea | 1 |
| Síndrome de Sweet | 1 |
| Vitiligo | 1 |
| Atividade de piodermite associada à atividade de comorbidade (n=40) | 6 (15,4) |
| Patergia | 14 |
| Enxerto | 1 |
| Hepatectomia | 1 |
| Biopsia de mama | 1 |
| Ferimento levea | 11 |
| Histórico familiar de doenças associadas | 3 (6,0) |
| Doença de Crohn | 2 |
| Colite ulcerativa | 1 |
| Comorbidades relacionadas ao tratamento | |
| Hipertensão arterial | 11 (22,0) |
| Sepse | 7 (14,0) |
| Diabetes | 5 (10,0) |
| Uso de substâncias | |
| Fumo | 14 (28) |
| Cocaína | 2 |
| Crack | 1 |
Patergia foi observada em 14 (28%) casos; em três deles após procedimentos médicos (biopsia de mama, hepatectomia e enxerto), e o restante após ferimentos leves.
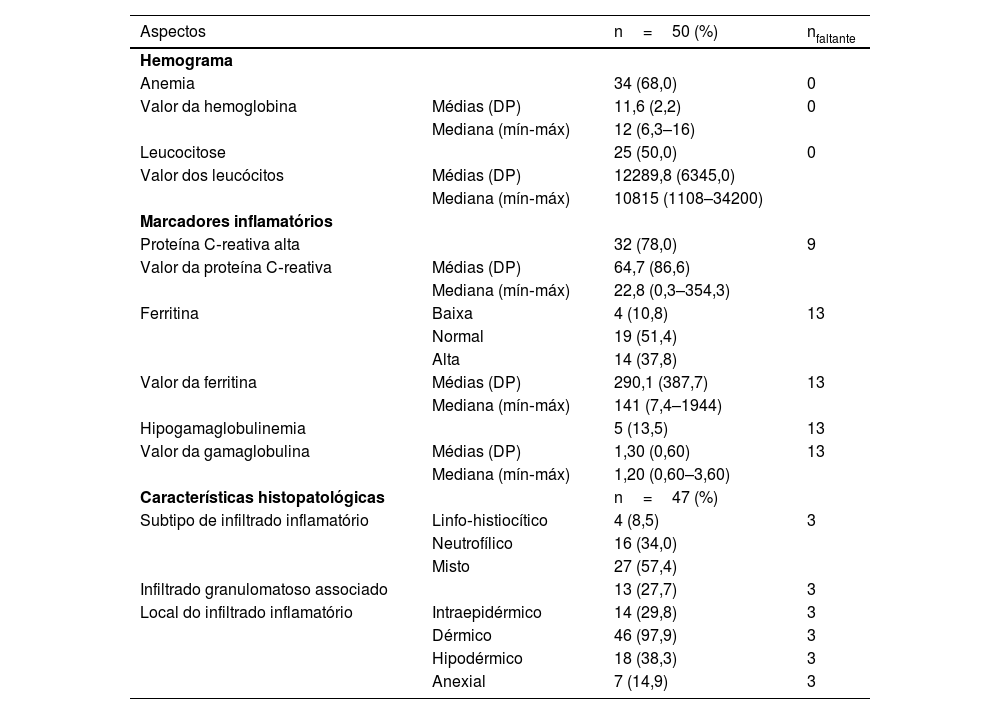
Testes laboratoriais e histopatológicosOs dados laboratoriais são mostrados na tabela 4.
Aspectos laboratoriais e histopatológicos
| Aspectos | n=50 (%) | nfaltante | |
|---|---|---|---|
| Hemograma | |||
| Anemia | 34 (68,0) | 0 | |
| Valor da hemoglobina | Médias (DP) | 11,6 (2,2) | 0 |
| Mediana (mín‐máx) | 12 (6,3–16) | ||
| Leucocitose | 25 (50,0) | 0 | |
| Valor dos leucócitos | Médias (DP) | 12289,8 (6345,0) | |
| Mediana (mín‐máx) | 10815 (1108–34200) | ||
| Marcadores inflamatórios | |||
| Proteína C‐reativa alta | 32 (78,0) | 9 | |
| Valor da proteína C‐reativa | Médias (DP) | 64,7 (86,6) | |
| Mediana (mín‐máx) | 22,8 (0,3–354,3) | ||
| Ferritina | Baixa | 4 (10,8) | 13 |
| Normal | 19 (51,4) | ||
| Alta | 14 (37,8) | ||
| Valor da ferritina | Médias (DP) | 290,1 (387,7) | 13 |
| Mediana (mín‐máx) | 141 (7,4–1944) | ||
| Hipogamaglobulinemia | 5 (13,5) | 13 | |
| Valor da gamaglobulina | Médias (DP) | 1,30 (0,60) | 13 |
| Mediana (mín‐máx) | 1,20 (0,60–3,60) | ||
| Características histopatológicas | n=47 (%) | ||
| Subtipo de infiltrado inflamatório | Linfo‐histiocítico | 4 (8,5) | 3 |
| Neutrofílico | 16 (34,0) | ||
| Misto | 27 (57,4) | ||
| Infiltrado granulomatoso associado | 13 (27,7) | 3 | |
| Local do infiltrado inflamatório | Intraepidérmico | 14 (29,8) | 3 |
| Dérmico | 46 (97,9) | 3 | |
| Hipodérmico | 18 (38,3) | 3 | |
| Anexial | 7 (14,9) | 3 |
DP, desvio padrão; mín, valor mínimo; máx, valor máximo.
Histopatologia estava disponível em 47 dos 50 casos, com predominância de infiltrado inflamatório misto (n=27/47; 57,4%) seguido por infiltrado neutrofílico (n=16/47; 34%), infiltrado granulomatoso (n=13/47; 37,7%) e infiltrado linfo‐histiocítico (n=4/47; 8,5%). A análise histopatológica detalhada está resumida na tabela 4.
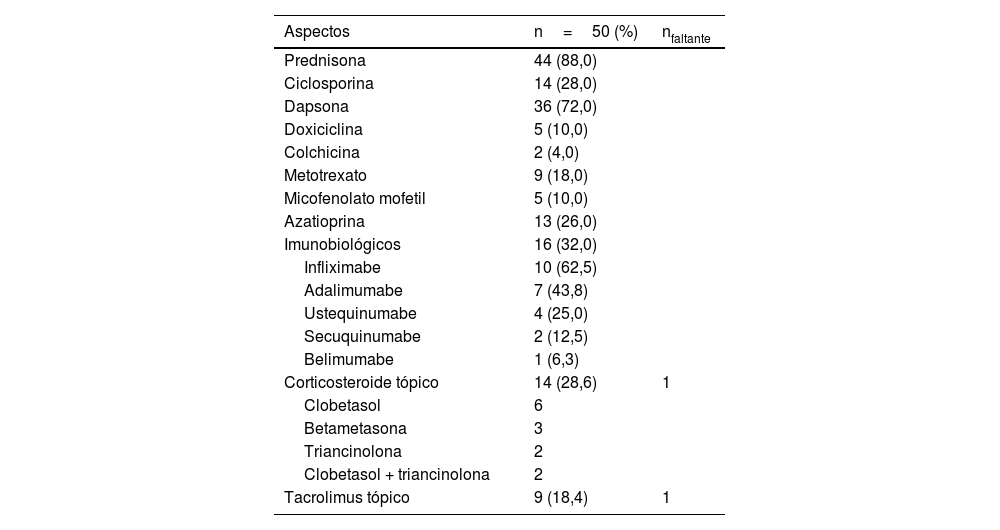
TratamentoPrednisona foi usada em 44 de 50 casos (88%), como medicamento único (n=7/50; 14%) ou em associação (n=37/50; 74%). O segundo medicamento mais utilizado foi a dapsona (n=36/50; 72%). Vale ressaltar que a dapsona sozinha resultou em controle completo da doença em dois casos. Quatorze pacientes (28%) utilizaram ciclosporina além de outros medicamentos, e 16 pacientes (32%) usaram pelo menos um agente imunobiológico (tabela 5). Entre esses 16 pacientes usando imunobiológicos, cinco obtiveram cura completa, tornando possível reduzir gradualmente e, eventualmente, suspender a prednisona e outros agentes. Em dez casos, o controle da doença foi obtido utilizando imunobiológicos em associação com outros imunossupressores clássicos (tabela 6). O controle não foi alcançado em um paciente com PG + HS, apesar da associação de alta dose de prednisona, ciclosporina e dois imunobiológicos diferentes (adalimumabe e ustequinumabe).
Medicação (isolada)
| Aspectos | n=50 (%) | nfaltante |
|---|---|---|
| Prednisona | 44 (88,0) | |
| Ciclosporina | 14 (28,0) | |
| Dapsona | 36 (72,0) | |
| Doxiciclina | 5 (10,0) | |
| Colchicina | 2 (4,0) | |
| Metotrexato | 9 (18,0) | |
| Micofenolato mofetil | 5 (10,0) | |
| Azatioprina | 13 (26,0) | |
| Imunobiológicos | 16 (32,0) | |
| Infliximabe | 10 (62,5) | |
| Adalimumabe | 7 (43,8) | |
| Ustequinumabe | 4 (25,0) | |
| Secuquinumabe | 2 (12,5) | |
| Belimumabe | 1 (6,3) | |
| Corticosteroide tópico | 14 (28,6) | 1 |
| Clobetasol | 6 | |
| Betametasona | 3 | |
| Triancinolona | 2 | |
| Clobetasol + triancinolona | 2 | |
| Tacrolimus tópico | 9 (18,4) | 1 |
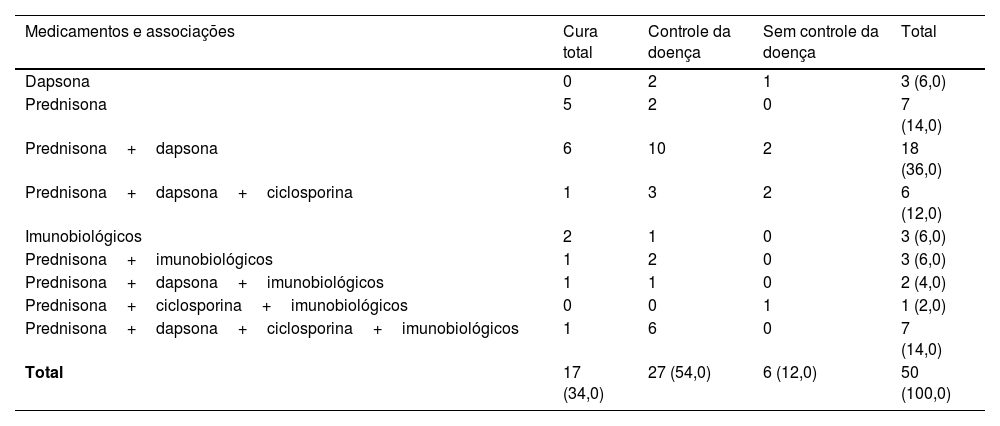
Medicamentos e associações
| Medicamentos e associações | Cura total | Controle da doença | Sem controle da doença | Total |
|---|---|---|---|---|
| Dapsona | 0 | 2 | 1 | 3 (6,0) |
| Prednisona | 5 | 2 | 0 | 7 (14,0) |
| Prednisona+dapsona | 6 | 10 | 2 | 18 (36,0) |
| Prednisona+dapsona+ciclosporina | 1 | 3 | 2 | 6 (12,0) |
| Imunobiológicos | 2 | 1 | 0 | 3 (6,0) |
| Prednisona+imunobiológicos | 1 | 2 | 0 | 3 (6,0) |
| Prednisona+dapsona+imunobiológicos | 1 | 1 | 0 | 2 (4,0) |
| Prednisona+ciclosporina+imunobiológicos | 0 | 0 | 1 | 1 (2,0) |
| Prednisona+dapsona+ciclosporina+imunobiológicos | 1 | 6 | 0 | 7 (14,0) |
| Total | 17 (34,0) | 27 (54,0) | 6 (12,0) | 50 (100,0) |
É digno de nota que, para sete pacientes, a principal indicação do imunobiológico foi PG; para sete pacientes, a principal indicação foi DII (cinco por colite ulcerativa [CU] e dois por doença de Crohn [DC]); para um paciente, artrite psoriásica e para um paciente, artrite lúpica (belimumabe). Detalhes sobre medicamentos, associações e resultados são descritos na tabela 6.
Hospitalização e progressãoHospitalização foi necessária em 32 de 50 casos (64%) para controlar PG, dor ou infecção.
Controle da doença (cicatrização de úlceras, permitindo redução da dose do medicamento principal, geralmente prednisona) foi alcançado em 44 de 50 (88%) pacientes após média de 3,56 meses (mediana 1–34).
Recorrência de PG foi observada em 49% dos casos, na maioria deles (70,8%) envolvendo o mesmo local da lesão inicial.
Remissão completa da doença ocorreu em 12 pacientes (24%), que estão sem terapia. Cinco pacientes necessitaram de imunobiológicos para manter o controle da doença após redução total dos imunossupressores clássicos. A mediana de tempo livre da doença foi de 37 meses (12 a 192 meses).
Os pacientes que utilizaram imunobiológicos ficaram livres da doença em 31,3% dos casos, enquanto os pacientes que não usaram imunobiológicos ficaram livres da doença em 35,3% dos casos.
DiscussãoPG pode ocorrer em qualquer idade, especialmente entre 20 e 50 anos, e predomina em mulheres.1 Na presente série, a média de idade no momento do diagnóstico foi de 33,7 anos e 58% dos pacientes eram mulheres, o que é consistente com a literatura.1–3 Oito (16%) pacientes tinham menos de 18 anos no momento do início da doença.
Na maioria dos casos (52%), PG não foi a hipótese inicial, levando a um tempo médio desde o início dos sintomas até o diagnóstico de 26,5 meses. Esses dados demonstram que o PG ainda representa desafio diagnóstico, possivelmente relacionado à raridade da doença e à multiplicidade de diagnósticos diferenciais sendo considerado diagnóstico de exclusão apesar de sua apresentação característica.
Dentre os subtipos clínicos, a forma ulcerativa é a mais comum1,3–6 e também foi a mais frequente na presente análise (86%). As lesões afetaram predominantemente os membros inferiores, corroborando estudos anteriores.1,4,5,8–10Um dos casos da presente série apresentou envolvimento pulmonar, o que foi descrito anteriormente em 42 relatos de casos.7–9 Patergia é descrita em 25% a 50% dos casos,3,5,11 e ocorreu em 24% dos pacientes da presente série.
A forma pós‐operatória de PG mostra menor associação com doenças sistêmicas em comparação com outros tipos de PG; os distúrbios hematológicos são os mais prevalentes. As regiões mamárias e abdominal são as mais comumente afetadas em casos pós‐operatórios e, em média, o acometimento ocorre em torno de sete dias após a cirurgia. Como é frequentemente diagnosticado erroneamente como ferida infectada, o desbridamento pode induzir a progressão das lesões. PG deve ser incluído no diagnóstico diferencial de deiscência de ferida pós‐operatória.26
Dor local, que geralmente é desproporcional ao tamanho da lesão, ocorreu em 78% dos casos da presente série, e 24% dos pacientes apresentaram febre.6 Binus et al. observaram dor em 62,1% dos casos em um estudo retrospectivo incluindo 103 pacientes com PG.6
Doenças sistêmicas, principalmente DII, artropatias inflamatórias e distúrbios hematológicos,6,12,13 podem preceder, coexistir ou seguir o diagnóstico de PG. A maioria dos pacientes da presente série (80%) tinha pelo menos uma condição associada; nessa série, a condição não dermatológica mais comum foi DII (20%). PG também pode ocorrer no contexto de síndrome paraneoplásica. De acordo com Shah et al., PG paraneoplásico geralmente se apresenta como lesão ulcerativa nas extremidades em associação com tumor de mama.15 Dois dos pacientes da presente série tinham histórico de neoplasia sólida (fígado e próstata).
Um paciente desenvolveu síndrome de Sweet e dez (20%) apresentavam HS. Seis (12%) dos pacientes da presente série tinham síndromes autoinflamatórias associadas (PASH, PAPASH, PAPA e doença autoinflamatória indefinida). A coexistência de PG e síndrome de Sweet foi relatada.19,20,28–33 Outras dermatoses observadas nos pacientes da presente série incluíram: acne conglobata, lúpus eritematoso (com artrite lúpica), psoríase (com artrite psoriásica), vitiligo, pustulose subcórnea e eritema anular centrífugo.
Na presente série, 17 pacientes relataram uso de tabaco (n=14), cocaína (n=1) e crack (n=1). Keith et al. relataram a associação entre PG e cocaína adulterada com levamisol. A cocaína tem efeito tóxico nas células endoteliais, e o levamisol promove vasculopatia, que são prováveis mecanismos fisiopatológicos no desenvolvimento de PG.21–23 Um paciente desenvolveu PG após tratamento de acne com isotretinoína e um paciente após terapia anti‐IL6 (tocilizumabe) para artrite reumatoide. Casos induzidos por medicamentos são incomuns e podem estar relacionados à migração e funcionalidade anormais de neutrófilos, desencadeando resposta inflamatória desregulada e apoptose de queratinócitos.19,21,22
Anemia ocorreu na maioria dos pacientes da presente série (68%) em decorrência de vários fatores, incluindo doenças associadas (DII, distúrbios hematológicos), cronicidade do PG, sangramento das lesões, e hemólise induzida por dapsona. Também foram observados leucocitose e aumento de proteína‐C reativa (PCR), o que pode ser atribuído ao estado pró‐inflamatório do PG, à associação frequente com infecção secundária e neutrofilia periférica induzida por corticosteroides sistêmicos.11,23
Infiltração neutrofílica acentuada estava presente em 34% dos casos do presente estudo, afetando também os anexos em 14,9%, e quatro dos casos da presente série (8,5%) apresentavam infiltrado linfo‐histiocítico. Infiltrado granulomatoso também pode ocorrer1 e foi observado em 27,7% dos casos na presente série. Poucos estudos avaliaram as características histopatológicas do PG. Chakiri et al. relataram que entre 14 pacientes com PG, infiltrado neutrofílico denso ocorreu em todos os casos, com vasculite em quatro casos e infiltrado linfoplasmocítico em cinco casos.1 Os achados histopatológicos do PG não são específicos e variam de acordo com o estágio da lesão. Lesões iniciais podem mostrar supuração na derme profunda, frequentemente foliculocêntrica, e infiltrados neutrofílicos densos.1,11 Após a ulceração, pode haver necrose e hemorragia, trombose de vasos sanguíneos dérmicos ou hipodérmicos, com infiltrado linfocítico.11
O tratamento do PG visa reduzir a atividade inflamatória e a dor, promover a cicatrização de feridas e controlar doenças associadas.2,11,24,25 Ele ainda representa um desafio, pois nenhuma terapia específica está disponível atualmente e não há consenso sobre qual tratamento é o mais eficaz. Os corticosteroides sistêmicos são geralmente eficazes e considerados o tratamento de primeira linha em dose equivalente a 0,5–1mg/kg/dia de prednisona. Lesões mais resistentes requerem terapia mais longa (> três meses), doses maiores ou associação com outros agentes imunomoduladores. No presente estudo, a maioria dos casos foi tratada com prednisona 0,25–1,5mg/kg/dia (88%) como terapia de primeira linha.1,2,4–6,11,24 A ciclosporina também é considerada terapia de primeira linha, frequentemente em associação com prednisona em casos recalcitrantes, e foi necessária em 28% dos casos do presente estudo em algum momento do seguimento.27
Outros imunossupressores e imunomoduladores podem ser indicados, como dapsona, azatioprina, micofenolato mofetil, ciclofosfamida e metotrexato. Agentes imunobiológicos, incluindo infliximabe, adalimumabe, ustequinumabe e canaquinumabe, também foram utilizados.1,2,4–6,11,24 O segundo medicamento mais utilizado na presente série foi a dapsona (72%), e dois em cada três pacientes obtiveram controle da doença com dapsona em monoterapia. Azatioprina também foi usada em 13 dos pacientes da presente série para o tratamento de DII; entretanto, o mesmo não induziu melhora das lesões de PG.1,2,4–6,11,24
Imunobiológicos são opções terapêuticas promissoras em virtude de sua atividade em citocinas inflamatórias envolvidas na patogênese de PG, como TNF‐α, IL‐1, e IL‐17.2,4,24,27,28 Dezesseis dos pacientes do presente estudo (32%) utilizaram pelo menos um agente imunobiológico, uma vez que as lesões de PG eram refratárias à terapia com corticosteroides (n=9) ou para o tratamento de comorbidade (n=7). Foi demonstrado que alguns pacientes com DII têm resposta adequada apenas com imunobiológicos, sugerindo que eles devem ser considerados terapia de primeira linha para o tratamento de PG.8,11,12
A maioria dos pacientes necessitou de hospitalização pelo menos uma vez (32 casos, 64%), semelhante à taxa relatada por Platzer et al. em uma revisão de 36 casos (69,4%).4 Sete (14%) dos pacientes do presente estudo desenvolveram sepse durante terapia imunossupressora em pelo menos uma das recorrências da doença. Desses sete pacientes, três usaram prednisona + dapsona + ciclosporina + imunobiológicos; um usou prednisona + dapsona + imunobiológicos; um usou prednisona + imunobiológicos; e dois usaram apenas prednisona. As infecções estão entre as principais causas de morte em estudos de PG.4,13 Dois dos pacientes do presente estudo apresentaram catarata grave como complicação do uso prolongado de prednisona. Um paciente desenvolveu nefropatia irreversível e doença desmielinizante após prednisona e ciclosporina. Esses dados podem sugerir benefícios da introdução precoce de imunobiológicos.
Controle da doença foi alcançado em 44 de 50 pacientes (88%) após média de 3,56 meses, consistente com as taxas de remissão descritas.3 Recorrência de PG ocorreu em 49% dos pacientes do presente estudo, principalmente no local anterior (70,8%). Taxas de recorrência variáveis foram relatadas na literatura (17%–61%) e também foram mais comuns no local da lesão primária.3,4
Ao final do seguimento dos pacientes do presente estudo, entre 17 pacientes (34%) que estavam livres da doença, cinco permaneceram usando imunobiológicos, enquanto 12 estavam sem qualquer terapia de manutenção. Todos os cinco pacientes que alcançaram remissão completa da doença com imunobiológicos tinham DII (três CU e dois DC) e três também apresentavam HS. Nenhum dos cinco teve recorrência das lesões. Esses dados fornecem evidências adicionais de que pacientes com comorbidades podem se beneficiar do uso precoce de imunobiológicos.8,11,12 Vale ressaltar que 20 de 32 (62,5%) pacientes com PG idiopática ou HS associada ou PG sindrômico foram controlados com imunossupressores clássicos, e seis (18,75%) deles só alcançaram o controle da doença com imunobiológicos.
As principais limitações do presente estudo são sua natureza retrospectiva, o desenho descritivo e o número de pacientes. Portanto, embora pareça uma amostra pequena quando consideradas doenças mais comuns, o PG é doença rara, e esta é uma das maiores séries já publicadas incluindo a América Latina e pacientes com longo seguimento.
ConclusãoO presente estudo confirma que PG é doença rara, frequentemente difícil de diagnosticar e associado a doenças sistêmicas e síndromes autoinflamatórias. Embora os corticosteroides sistêmicos permaneçam como terapia de primeira linha, os dados do presente estudo sugerem que expandir o uso de imunobiológicos no início do tratamento de PG pode representar ferramenta terapêutica promissora para evitar recorrências da doença e hospitalizações frequentes.
Suporte financeiroNenhum.
Contribuição dos autoresLivia Maria Oliveira Salviano: Aprovação da versão final do manuscrito; revisão crítica da literatura; obtenção, análise e interpretação dos dados; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; elaboração e redação do manuscrito; análise estatística; concepção e planejamento do estudo.
Denise Miyamoto: Aprovação da versão final do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; concepção e planejamento do estudo.
Claudia Giuli Santi: Aprovação da versão final do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; concepção e planejamento do estudo.
Tatiana Mina Yendo: Aprovação da versão final do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; concepção e planejamento do estudo.
Maria Cecilia Rivitti‐Machado: Aprovação da versão final do manuscrito; participação efetiva na orientação da pesquisa; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados; revisão crítica do manuscrito; concepção e planejamento do estudo.
Conflito de interessesNenhum.
Como citar este artigo: Salviano LM, Miyamoto D, Santi CG, Yendo TM, Rivitti‐Machado MC. Pyoderma gangrenosum: a 22‐year follow‐up of patients in a tertiary reference hospital in Brazil. An Bras Dermatol. 2025;100:462–9.
Trabalho realizado no Hospital das Clínicas, Faculdade de Medicina, Universidade de São Paulo, São Paulo, SP, Brasil.
