

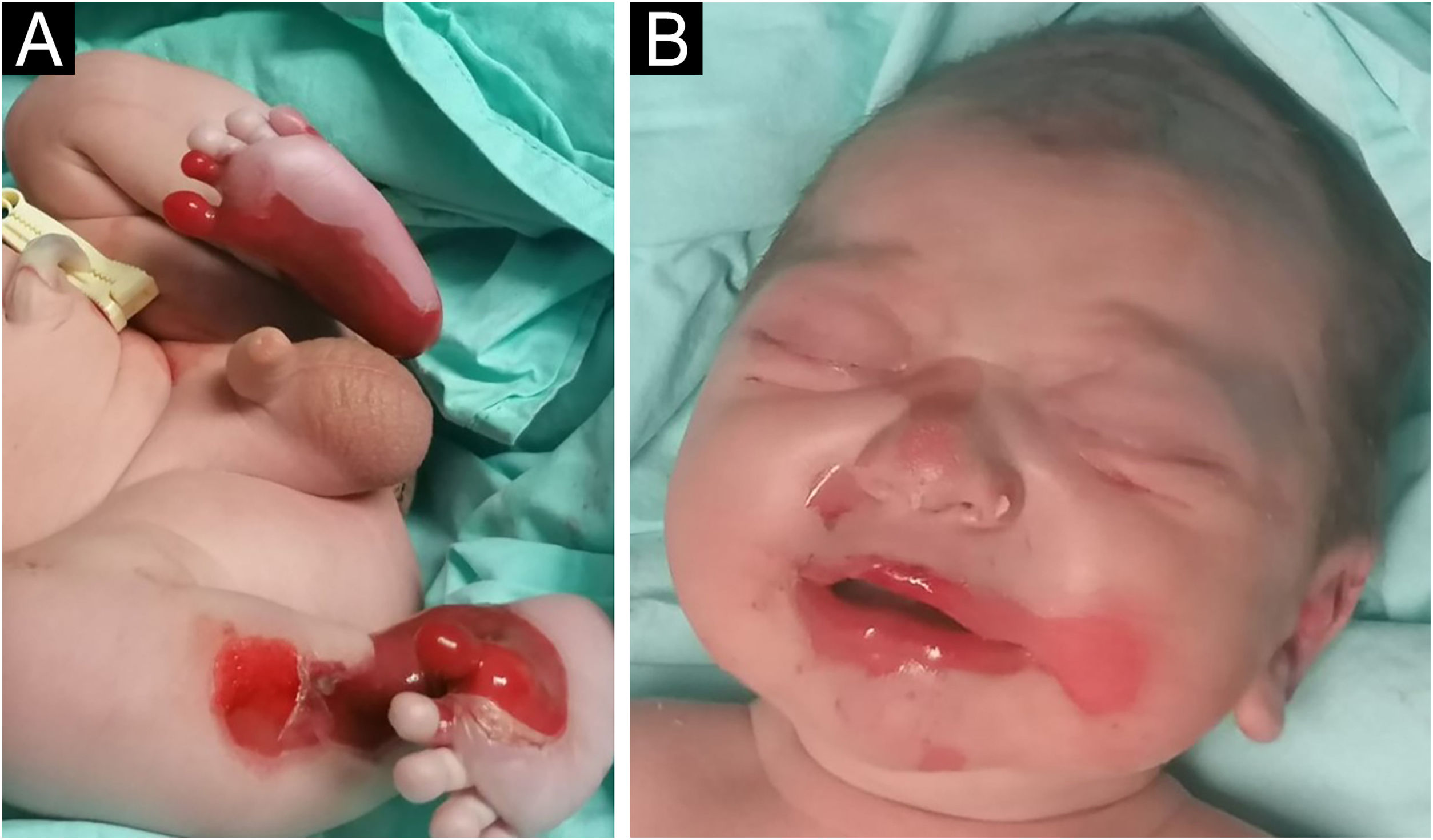
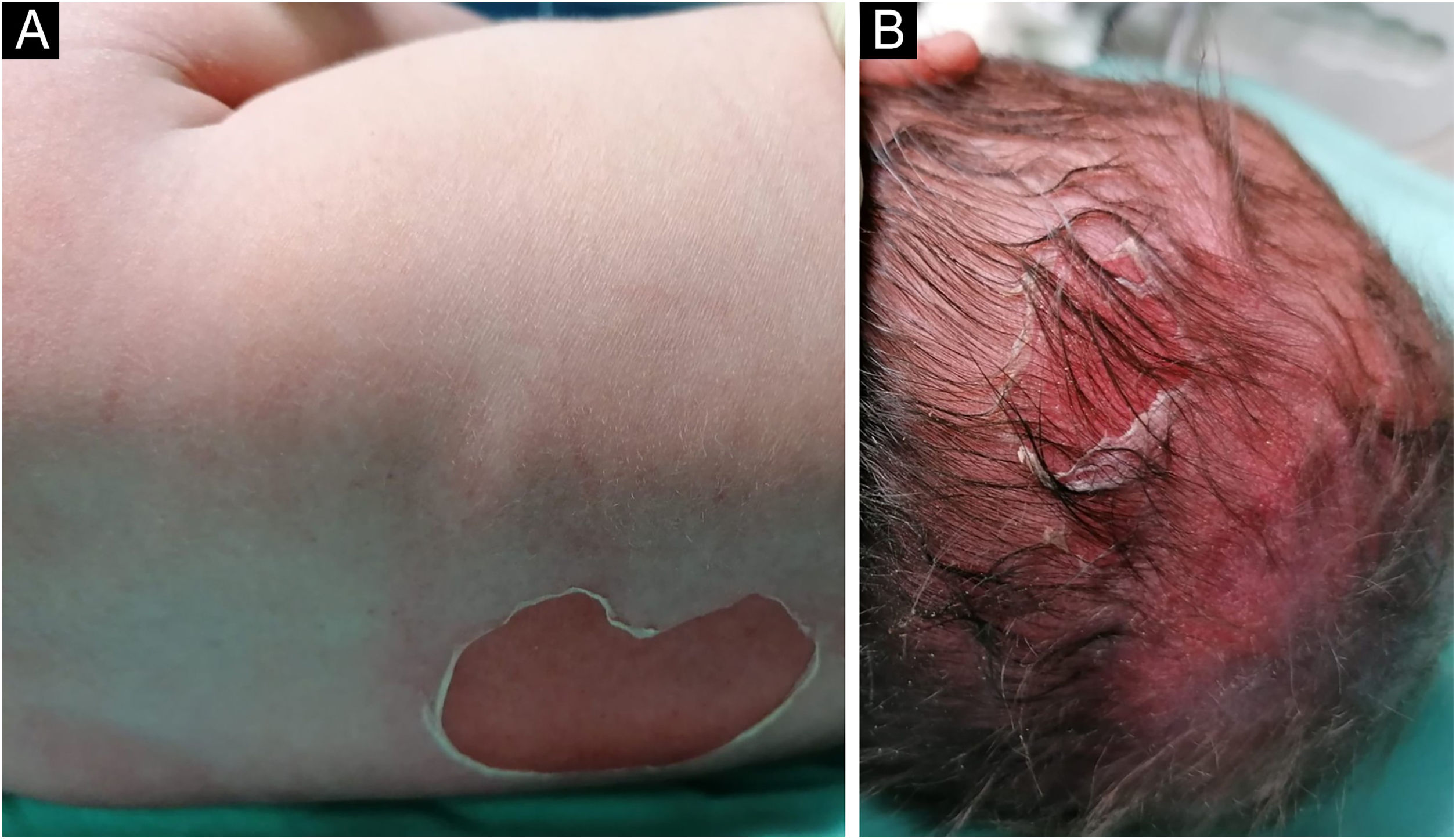
Lactente do sexo masculino, nascido a termo, apresentou ao nascimento bolhas e diversas áreas com erosão, principalmente nos pés e na face, mas também afetando o tronco, o couro cabeludo e a região perineal (figs. 1 e 2). Apresentava, ainda, opacidade da córnea direita com hipervascularização e hemorragia. Unhas, cabelos e mucosas eram normais. O paciente era o primeiro filho de um casal saudável, não consanguíneo e sem histórico familiar de dermatoses bolhosas. Um painel de 22 genes realizado por sequenciamento de próxima geração (NGS, do inglês next‐generation sequencing) encontrou variante patogênica, c.325_326insCG (p.Glu109Alafs*39), em homozigose no gene COL7A1. Nos primeiros meses de vida, o quadro piorou com novas lesões nas mãos e axilas, além de cicatrizes mutilantes nos pés. Infelizmente, os pais recusaram estudos adicionais. Com base na apresentação clínica e na análise genética, foi feito o diagnóstico de epidermólise bolhosa distrófica (EBD) recessiva grave.
 e na região perioral (B).")
 e couro cabeludo (B).")
A EBD é doença hereditária de fragilidade da pele, caracterizada pela formação de bolhas na sublâmina densa. É causada por variantes patogênicas no gene COL7A1, que codifica o colágeno tipo VII, responsável pela coesão da junção dermoepidérmica. O padrão de herança pode ser recessivo (EBDR) ou dominante (EBDD).1,2 O espectro clínico é altamente variável, incluindo desde formas bolhosas generalizadas, com envolvimento mucoso, até formas localizadas leves; em geral, as formas recessivas são mais graves.2 Essas últimas geralmente estão associadas a variantes que causam códons de parada prematura (PTC, do inglês premature stop codons), com posterior síntese proteica truncada. Em heterozigosidade, as variantes PTC não causam a doença, mas heterozigosidade composta com variantes missense pode estar associada a formas mais leves de EBDR. Os casos de EBDD são geralmente decorrentes de substituições de glicina, mas outras mutações podem estar envolvidas. As substituições de glicina também podem ser herdadas recessivamente, e algumas substituições específicas foram associadas tanto à EBDR quanto à EBDD.1‐3
A gravidade dos sintomas depende do nível de expressão de COL7A1, que é determinado pelo tipo e posição da variante patogênica.1‐3 Entretanto, a correlação genótipo‐fenótipo não é consistente, e variantes idênticas podem resultar em fenótipos diferentes, o que sugere que outros fatores, genéticos ou ambientais, podem estar envolvidos nessa divergência fenotípica.1,4
A variante apresentada pelo presente caso é rara, com menos de 10 casos relatados anteriormente. Curiosamente, essa variante só foi relatada no norte de Portugal e no Brasil, o que pode ser decorrente de efeito fundador, relacionado com a antiga colonização portuguesa.1
O presente caso contribui para o espectro mutacional associado à EBDR e para a melhor compreensão de sua correlação clínica.
Suporte financeiroNenhum.
Contribuição dos autoresPatrícia Amoedo: Redação e edição (principal).
Ana Grangeia: Revisão (suporte).
Lígia Peralta: Revisão (suporte).
Alberto Mota: Revisão e aprovação final.
Conflito de interessesNenhum.
Como citar este artigo: Amoedo P, Grangeia A, Peralta L, Mota A. A case of dystrophic epidermolysis bullosa with a rare COL7A1 variant. An Bras Dermatol. 2024;99:448–9.
Trabalho realizado no Centro Hospitalar e Universitário de São João, Porto, Porto, Portugal.

- Pitiríase rubra pilar após vacinação contra COVID‐19: tratamento bem‐sucedido com ustequinumabe
- A imunoexpressão da proteína 8‐hidroxi‐2’‐desoxiguanosina está associada à patogênese da queilite actínica
- Caso de dermatofibroma atrófico: possível papel da metaloproteinase de matriz‐2
- Dermatomiosite bolhosa com anticorpos anti‐NPX‐2 associada a câncer de mama