






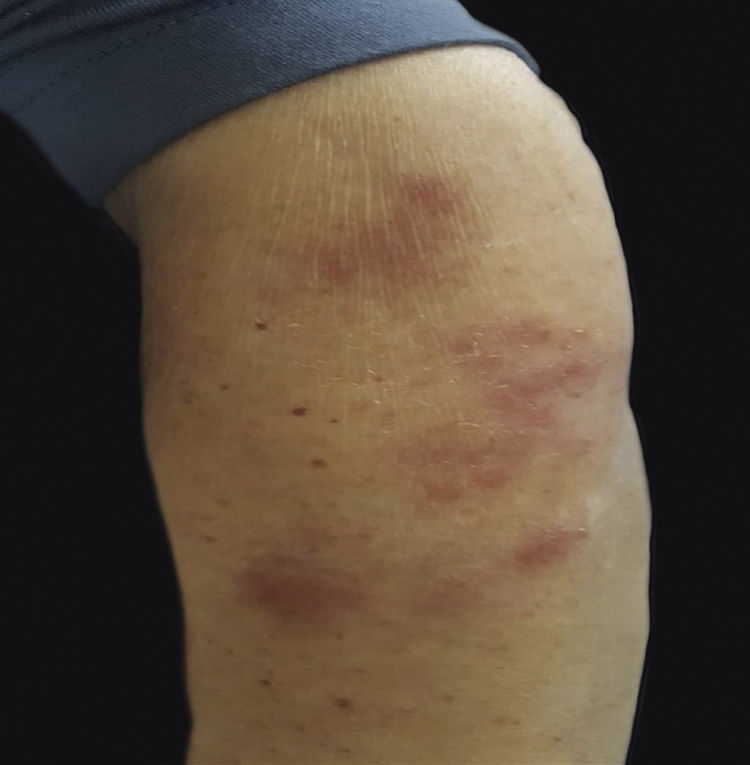
Uma mulher de 76 anos apresentava várias placas arroxeadas nos braços (fig. 1), na região deltoide, nos cotovelos, no punho, na região mamária e nas pernas. As lesões eram dolorosas ao toque, mas sem prurido. Clinicamente, havia suspeita de eczema. Foi solicitada uma biópsia de pele do braço esquerdo.
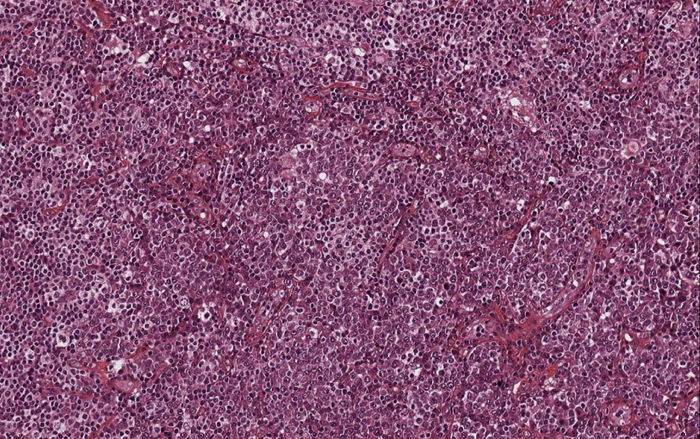
O exame histológico das seções coradas com hematoxilina e eosina indicou presença de aglomerados difusos de células atípicas em forma oval, com citoplasma abundante, núcleos excêntricos, cromatina com aspecto de mostrador do relógio e, às vezes, nucléolos proeminentes, infiltravam‐se na derme média e profunda (fig. 2). Figuras mitóticas foram observadas. As células neoplásicas eram morfologicamente semelhantes às células plasmáticas maduras. Portanto, foi feito um painel imuno‐histoquímico específico: elas eram difusamente positivas para CD79a, CD138, CD56, MUM‐1 e EMA e totalmente negativas para CD20. Estudos imuno‐histoquímicos para cadeias leves kappa e lambda evidenciaram expressão monoclonal de cadeias leves de imunoglobulina kappa (fig. 3).

Para completar o processo diagnóstico, foi feita uma biópsia da medula óssea, negativa para localização de mieloma múltiplo (MM) – menos de 10% de células plasmáticas; sem restrição clonal. Não foram observadas proteínas Bence‐Jones na urina. O hemograma e a análise bioquímica do sangue revelaram valores normais de hemoglobina, creatinina sérica e cálcio sérico. A eletroforese de proteínas séricas evidenciou um pico da cadeia leve lambda.
Uma vez confirmada a ausência de doença em outras localizações, foi feito o diagnóstico clínico‐patológico de plasmocitoma cutâneo difuso primário (PCDP). Considerando o amplo comprometimento cutâneo, a paciente foi submetida a um tratamento sistêmico: bortezomibe na dose de 1,3mg/m2 por via subcutânea nos dias 1, 8, 15 e 22, melfalano administrado por via oral na dose de 14mg nos dias 1, 2, 3 e 4 e dexametasona na dose de 20mg nos dias 1, 2, 8, 9, 15, 16, 22, e 23 (VMP). Após nove ciclos, a tomografia computadorizada por emissão de pósitrons com fluorodeoxiglucose evidenciou o desaparecimento completo das lesões de pele e ausência do pico da imunoglobulina G lambda na eletroforese de proteínas séricas.
A paciente completou o tratamento sem efeitos adversos e, até o momento, após um ano e oito meses de acompanhamento, nenhuma recorrência foi detectada.
O PCDP é uma doença rara1 que surge primariamente na pele; portanto, pode ser considerado um plasmocitoma cutâneo extramedular localizado (PEM) e não deve ser confundido com plasmocitoma cutâneo secundário (PCS) no contexto de MM.2 De acordo com uma revisão sistemática recente, apenas 68 casos de plasmocitomas cutâneos primários (PCP) foram relatados na literatura, a maioria dos quais eram lesões isoladas.3
O PCDP geralmente surge como nódulos cutâneos azul‐arroxeados com predileção por face, tronco e extremidades.1,2 Histologicamente, um padrão de infiltração difusa ou nodular pode ser observado nos PCP; as células neoplásicas podem apresentar diferentes estágios do processo de maturação das células plasmáticas, desde características bem diferenciadas a características pleomórficas (semelhantes aos plasmoblastos).4,5 PCPs com características semelhantes a plasmoblastos são compostos de células neoplásicas com maior razão nuclear/citoplasmática, cromatina finamente dispersa e nucléolos mais proeminentes.4 O epidermotropismo geralmente está ausente nas PCPs.5
O principal fator prognóstico é a apresentação clínica (lesões únicas ou múltiplas),3 mas também é importante considerar o desempenho do paciente e as comorbidades que podem prejudicar a adesão ao tratamento. Em sua revisão sistemática, Tsang et al.3 mostraram que a única variável clínica associada à sobrevida livre de recorrência (SLR) e à sobrevida global (SG) foi o número de lesões (únicas ou múltiplas); em particular, esses autores observaram uma grande diferença na sobrevida mediana e na SLR entre pacientes com lesões únicas e aqueles com lesões múltiplas.3 Nesse último subconjunto de pacientes, quimioterapia sistêmica é necessária devido ao alto índice de evolução para MM e ao mau prognóstico.1
O diagnóstico diferencial de PCP inclui MM com PCS, plasmocitoma extramedular com envolvimento cutâneo secundário, outros linfomas cutâneos de células B, em particular linfoma de zona marginal (LZM) com diferenciação acentuada de células plasmáticas e doenças infecciosas como infecções por Borrelia.
É fundamental enfatizar que as células plasmáticas neoplásicas nas PCPs podem ser citologicamente indistinguíveis das reativas em doenças infecciosas, o que pode ser uma possível armadilha diagnóstica para os patologistas. Portanto, a avaliação imuno‐histoquímica da expressão mono ou policlonal das cadeias leves de imunoglobulina,4 combinada com a ausência de histórico sugestivo de infecção ou identificação de agentes causais, é essencial para o diagnóstico de malignidade.5
O MM raramente envolve a pele e, devido à ausência de características histológicas distintivas, a distinção entre SCP em MM e PCP só é possível por meio de exames clínicos e laboratoriais.2
Finalmente, o PCDP é uma doença rara que requer uma ampla abordagem multidisciplinar, fortemente recomendada para se obter o diagnóstico de PCP “verdadeiro” e assim escolher o tratamento ideal.
Suporte financeiroNenhum.
Contribuições dos autoresGiuseppe Broggi: Aprovação da versão final do manuscrito; concepção e planejamento do estudo; elaboração e redação do manuscrito.
Enrica Martino: Concepção e planejamento do estudo; elaboração e redação do manuscrito.
Valeria Calafiore: Obtenção, análise e interpretação dos dados; participação intelectual em conduta propedêutica e/ou terapêutica de casos estudados.
Rosario Caltabiano: Aprovação da versão final do manuscrito; revisão crítica do manuscrito.
Conflitos de interesseNenhum.
Como citar este artigo: Broggi G, Martino E, Calafiore V, Caltabiano R. Primary diffuse cutaneous plasmacytoma: when a correct clinico‐pathologic approach is mandatory for the patient's health. An Bras Dermatol. 2019;94:767–9.
Trabalho realizado no AOU Policlinico‐Vittorio Emanuele, Catania, Itália.
