






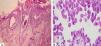

Pemphigus vulgaris is a chronic autoimmune bullous dermatosis that results from the production of autoantibodies against desmogleins 1 and 3. It is the most frequent and most severe form of pemphigus, occurring universally, usually between 40 and 60 years of age. It usually begins with blisters and erosions on the oral mucosa, followed by lesions on other mucous membranes and flaccid blisters on the skin, which can be disseminated. There is a clinical variant, pemphigus vegetans, which is characterized by the presence of vegetating lesions in the large folds of the skin. Clinical suspicion can be confirmed by cytological examination, histopathological examination, and direct and indirect immunofluorescence tests. The treatment is performed with systemic corticosteroids, and immunosuppressive drugs may be associated, among them azathioprine and mycophenolate mofetil. More severe cases may benefit from corticosteroids in the form of intravenous pulse therapy, and recent studies have shown a beneficial effect of rituximab, an anti-CD20 immunobiological drug. It is a chronic disease with mortality around 10%, and septicemia is the main cause of death. Patients need long-term and multidisciplinary follow-up.
Phemphigus diseases are a group of rare autoimmune bullous diseases that affects the skin and mucous membranes. Their estimated incidence is two new cases/million inhabitants/year in central Europe. They present chronic evolution, with significant morbidity and mortality, as well as an important impairment in quality of life.1,2 They originate from the production of pathogenic autoantibodies (usually of the IgG class) directed against different proteins of desmosomes (desmogleins). The union of these autoantibodies to the components of the desmosomes compromises intraepi-dermal adhesion, leading to acantholysis and formation of vesicles, blisters, and erosions on the skin and/or mucous membranes.3–5
Different types of pemphigus have been identified based on the clinical and histopathological characteristics, as well as on the specific antigens against which the autoantibodies are produced. The main forms are pemphigus vulgaris (PV) and pemphigus foliaceus (PF), but in the last decades non-classical forms of pemphigus have also been described: paraneoplastic pemphigus, pemphigus herpetiformis, and IgA pemphigus.6
The formation of autoantibodies against components of desmosomes has long been considered the main process in pemphigus pathogenesis. In addition to the important role of humoral immunity, cellular immunity has also been highlighted in the literature.7 PV is the main clinical form of pemphigus, accounting for approximately 70% of cases; it is also considered the most severe form of the disease.
EpidemiologyDespite being observed worldwide, the distribution of PV is ethnically and geographically unequal. Its incidence ranges from 0.76 new cases per million/year in Finland and 3.5 new cases per million/year in Japan to 16.1 cases per million/year in Jerusalem - worldwide, the incidence of this disease is highest in Ashkenazi Jews of Mediterranean origin.8 In most countries, PV is more frequent than PF - in France, for example, PV accounts for 73% of cases of pemphigus, and in Japan the ratio between PV and PF is 2:1. The exceptions are Finland, Brazil, and Tunisia; in the latter two, there are endemic foci of PF.9 In Brazil, endemic foci of PV are suspected in the central-west (Brasilia, DF) and southeast regions (Ribeirão Preto, SP). Studies present conflicting data regarding the evolution of PV incidence: while in Brazil and in the United Kingdom the incidence has increased in the last decade, in Israel a reduction was observed over the last 16 years.8–10
Similarly to other autoimmune diseases, PV is more prevalent among women. The male/female ratio ranges from 1:1.5 in Israel and Iran to 1:4 in Tunisia. PV may occur at any age, and disease onset is usually between 40 and 60 years of age. An increased frequency in the elderly and children has been observed. Interestingly, in some countries of the Middle East and Brazil, disease onset is earlier: a Brazilian study estimated that 17.7% of cases occur before the age of 30 years.8–10
Comorbidities and AssociationsThe influence of genetic and immunological factors on PV onset is well established. However, environmental factors (such as drugs, diet, and viruses, among others) may induce or impact the disease.11 Surprisingly, a recent systematic review concluded that smoking is a possible protective factor for PV, although other studies with different methodologies have failed to replicate this result.12
Recently, new associations between PV and various conditions have been described in adults and children, including infections and autoimmune, cardiovascular, endocrine, hematological, and neuropsychiatric diseases. The disorders most strongly associated with PV were myasthenia gravis, mucositis, insomnia, hidradenitis, and hematological neoplasias.2 The relationship between PV and cardiovascular diseases attributed to the chronic inflammatory process, reduction of physical activity due to pain, and discomfort caused by the lesions and use of systemic corticosteroids were also demonstrated. While its pathophysiology remains uncertain, it is crucial to assess the cardiovascular risk of patients with PV.13 Individuals with autoimmune diseases tend to develop autoimmune comorbidities. In cohort studies, PV has been associated with systemic lupus erythematosus, rheumatoid arthritis, autoimmune thyroiditis, type 1 diabetes, and myasthenia gravis.14 It is also important to assess the mental health of PV patients, since higher rates of depression were observed in all age groups, including children, as well as Parkinson's disease.15
Although the relationship between hematological neoplasias and paraneoplastic pemphigus is indisputable, there is still a lack of evidence to prove the association with PV.16 An uncontrolled study demonstrated that the frequency of non-Hodgkin's lymphoma and leukemias in PV cases was 50% higher than expected.2 A recent German study suggested that the prevalence of hematological neoplasias is twice as high in patients with PV when compared with controls.17 Finally, a population study that included 1,985 patients with pemphigus and 9,874 controls found that the prevalence of chronic leukemias, multiple myelomas, and non-Hodgkin's lymphomas was higher among PV cases when compared with controls. The association with chronic leukemia remained significant even after adjustment for PV immunosuppressive therapy.18 The following are possible explanations for the association between pemphigus and hematologic neoplasias: chronic immune stimulation and intense inflammatory process inducing pro-oncogenic mutations in cells in constant replication; persistent activation of B lymphocytes causing alteration in the cytokine profile and resistance to apoptosis; and the use of immunosuppressants for the treatment of PV, such as azathioprine, which increase the risk of hematological neoplasia.19
However, the relationship between PV and non-hematological neoplasia (i.e., solid organ neoplasia) is still incipient. Case-control studies have demonstrated an association between PV and oropharyngeal, gastrointestinal, and lung neoplasms.17,20 Recently, another branch of the population study that included 1,985 patients with pemphigus demonstrated a significant association with esophageal and laryngeal neoplasias, with prevalences three and two times higher than controls, respectively. No association was observed with other solid organ neoplasms. The following are possible explanations for the association between pemphigus and esophageal and laryngeal neoplasms: involvement of the mucous membranes of these organs in cases of PV, since they express desmoglein 3, the main PV antigen; and persistent inflammation, inducing mutations, resistance to apoptosis, and angiogenesis. Multivariate analysis ruled out the possibility of neoplasia secondary to the use of immunosuppressant as a treatment for PV.21
EtiopathogenesisAntigens in PVThe antigens involved in PV are desmogleins 1 (Dsg1) and 3 (Dsg3), which are 160 and 130 kDa transmembrane glycoproteins, respectively; they are an integral part of the desmosomes of the cadherin family, responsible for the intercellular adhesion of the squamous stratified epithelium. The basic pathophysiology of pemphigus is the inhibition of the adhesive function of desmogleins by autoantibodies, which leads to the formation of blisters.22
Cadherins are calcium-dependent intercellular adhesion molecules that are essential for tissue integrity. They can be divided into two groups: classic (cadherins P and N) and desmosomal caderins (desmogleins and desmocollins). Structurally, they have five extracellular domains that are indistinguishable between the groups, a transmembrane domain, and cytoplasmic domains that differ between classical and desmosomal cadherins. The distal extracellular domain of the cadherin molecule binds to the corresponding distal domain of the adjacent cell molecule; these are targeted by the autoantibodies responsible for cleavage (Figures 1 and 2).23
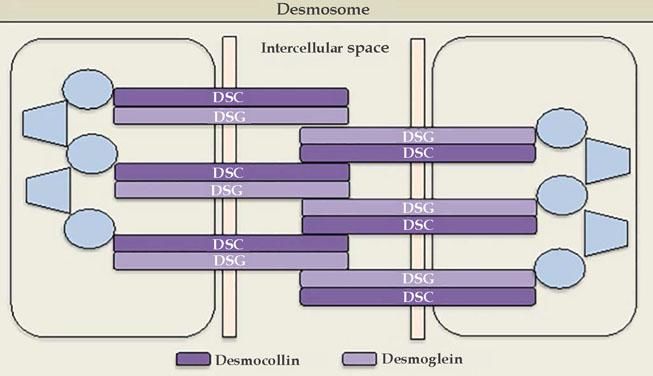
The desmosomal complex includes desmoglein, desmocollin, and transmembrane and cytoplasmic components
Source adapted from: Bolognia et al., 2018.9
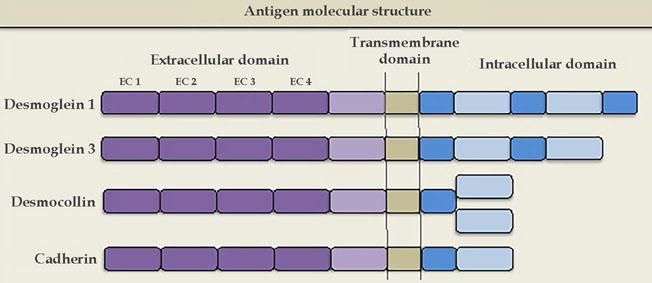
The molecular structure of the pemphigus antigens comprises the extracellular region (EC), with four calcium-dependent cadherin repeats
Source adapted from: Bolognia et al., 2018.9
Desmogleins have four isoforms: Dsg1 (160 kDa) and Dsg3 (130 kDa) are only expressed in the squamous stratified epithelium, where pemphigus bullous lesions occur; Dsg2 is expressed in all tissues with desmosomes, including the simple epithelium and the myocardium; Dsg4 is expressed in the hair follicles, and is possibly implicated in scalp lesions, which are common in pemphigus.24 Desmocollins are another group of transmembrane glycoproteins that, together with desmogleins, comprise the desmosome. It is not yet known whether desmocollins play a role in the etiopathogenesis of pemphigus and why they fail to compensate for the loss of desmoglein function.
Desmoglein compensation theoryIn 1999, Amagai & Stanley proposed the desmoglein compensation theory: Dsg1 and Dsg3 are compensated when coexpressed in the same cell, and the presence of one type of Dsg is sufficient to maintain the integrity of the skin or mucosa. This theory was based on the difference in the distribution of Dsg1 and Dsg3 between skin and mucosa - in the skin, Dsg1 is expressed throughout the epidermis, more intensely in the superficial layers, whereas Dsg3 is concentrated in the lower layers of the epidermis (basal and parabasal), rather than being expressed in the superficial epidermis; in the mucosa, Dsg1 and 3 are expressed, but Dsg1 is much smaller concentrations than Dsg3.5
Dsg1 is known to be the main PF antigen. In the skin, in cases of Dsg1 dysfunction, there is cleavage in the superficial epidermis, as this is the only region of the skin where Dsg1 is present alone, without Dsg3 co-expression. The deeper layers are not affected, as the presence of Dsg3 compensates for the dysfunction of Dsg1. In the mucosa, Dsg1 dysfunction does not lead to cleavage due to the co-expression of Dsg3 at higher concentrations throughout the epithelium extension. Thus, the predominant clinical picture is superficial lesions on the skin without mucosal involvement.
In mucosal PV, the main antigen involved is Dsg3. In the skin, isolated Dsg3 dysfunction is unable to produce blisters, as it is entirely compensated by Dsg1. However, in the mucosa, the low concentration of Dsg1 is not sufficient to compensate for the Dsg3 dysfunction, which leads to the predominance of mucosal lesions without cutaneous involvement.
In mucocutaneous PV, both Dsg1 and Dsg3 are involved. Therefore, there is extensive formation of blisters throughout the skin and mucous membranes. It is not yet clear why cleavage occurs only in the suprabasal layer and not throughout the epithelium, considering the expression of Dsg1 and Dsg3 throughout the epidermis. The following are possible explanations: antibodies from the dermis have easier access to the basal layer; and the intercellular adhesion of the basal layer may be weaker than that of the surface of the epidermis, due to the lower desmosome count (Figure 3).25
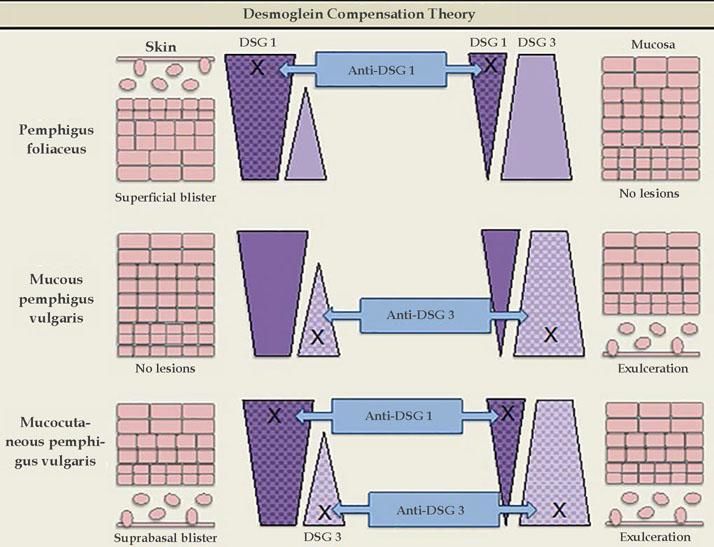
Desmoglein compensation theory. The different distribution patterns of Dsg1 and Dsg3 in the skin and mucosa are represented. In pemphigus foliaceus, anti-Dsg1 IgG antibodies cause superficial blisters on the skin, as Dsg3 compensates for non-functioning Dsg1 in the deep epidermis; there are no mucosal lesions, since adhesion is mediated mainly by Dsg3. In mucosal pemphigus vulgar-is, anti-Dsg3 IgG antibodies do not cause skin damage because Dsg1 compensates for Dsg3 dysfunction; however, there is a mucosal lesion because, unlike the skin, the low concentration of Dsg1 in the mucous membranes is not enough to compensate for Dsg3 dysfunction. In mucocutaneous pemphigus, the presence of anti-Dsg1 and anti-Dsg3 IgG antibodies causes lesions on both the skin and the mucosa
PV is caused by the presence of IgG autoantibodies directed against Dsg1 and/or Dsg3, part of the desmosomes present on the surface of keratinocytes, which play a primary pathogenic role in the induction of loss of intercellular adhesion, resulting in the formation of blisters. PV patients with only Dsg3 autoantibodies present the mucosal form of the disease. In turn, patients with DSg1 and Dsg3 autoantibodies have the mucocutaneous form of PV.
It has been demonstrated that the IgG4 subclass predominates in the acute phase of the disease, whereas IgG1 is associated with periods of remission. The possible involvement of IgM in the pathogenesis of PV is also being investigated, as well as the existence of autoantibodies against other antigens besides Dsg, such as desmocollins, plaquins, and mitochondria, among others.5,26
However, the mechanism by which the binding of autoantibodies to desmogleins leads to acantholysis remains uncertain. Possible hypotheses for this phenomenon include: alterations in in-tracellular transduction signaling and rupture of the cytoskeleton, resulting in keratinocyte shrinkage; spatial impediment for desmoglein adhesion; and formation of desmoglein-deficient desmosomes, among others.5
Furthermore, it is not clear what triggers pathogenic autoantibody production. It is known that there is a genetic predisposition determined by human leukocyte antigen (HLA); HLA--DRB1*04 and HLA-A*10 are more frequent in Ashkenazi Jews with pemphigus. A study in the Brazilian population showed the presence of DRB1*04:02 and DBQ1*05:03 alleles, and for the first time HLA-B*57 associated with PV.27
Clinical PictureThe clinical manifestation of PV may present mucosal or mucocutaneous involvement. Nearly all patients present mucosal lesions, mainly in the oral mucosa, with or without cutaneous lesions.
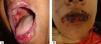
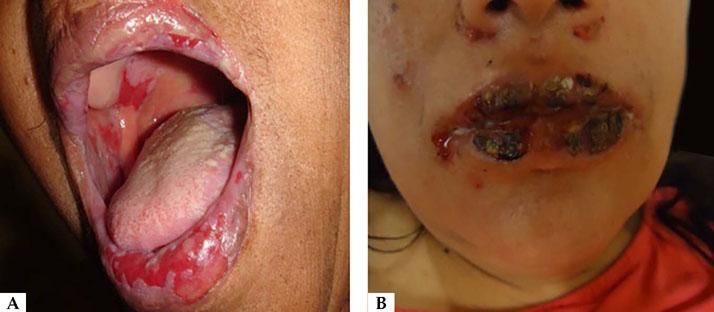
Oral lesions are the first manifestation in 50%–70% of cases and occur in 90% of patients during the course of the disease.28 They are characterized by painful erosions; blisters are rarely intact, probably because they are fragile and break easily. The most affected areas are the buccal and palatine mucosa, lips, and gingivae. The erosions are multiple and present in different sizes and irregular shapes; they extend peripherally and there is usually a delay in re-epithelization (Figure 4A).29 Gingival involvement manifests mainly as desquamative gingivitis.30 The lesions may extend to the vermilion border of the lips, forming a fissured hemorrhagic crust (Figure 4B).31 Oral lesions make feeding difficult, impairing the general and nutritional status. Other mucous membranes may be involved, including the conjunctiva, nasal mucosa, pharynx, larynx, esophagus, vagina, penis, and anus.31 Oral involvement may persist for months before progressing to involvement of the skin or other mucous membranes; it may also be the only manifestation of the disease.
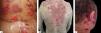
Cutaneous involvement can be localized or generalized. Most patients develop flaccid blisters of clear content on normal or erythematous skin. The blisters break easily, resulting in painful erosions that bleed easily (Figure 5A). Skin lesions can be observed in any location, but there is a predilection for the trunk, groin, armpits, scalp, and face; the palms and soles are usually spared. These erosions become covered by crusts, with no tendency to heal (Figure 5B).28,31,32 Healing is usually without a scar, but pigmentary changes may be observed.
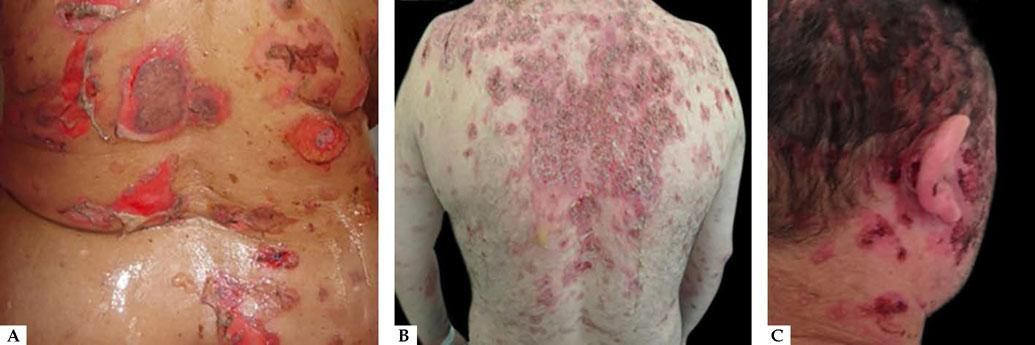
Due to the abundance of desmogleins in the hair follicle, the scalp is commonly affected in PV. Erosions, crusts, and scaly plaques can be observed, which may progress to alopecia (Figure 5C).33
Nikolsky's sign, characterized by the epidermal detachment caused by mechanical pressure at the edge of a blister or normal skin, is usually present in PV. Blisters can also be extended by vertical pressure over an intact blister, called the Asboe-Hansen sign or Nikolsky II sign.34 These signs clinically represent acantholysis or loss of cell adhesion and are not specific for PV; they may be present in other forms of pemphigus and in toxic epidermal necrolysis.
PV is a chronic disease, with periods of remission and exacerbation. Without proper treatment, PV can be fatal, as an extensive area of skin can lose its epidermal barrier function, leading to loss of body fluids, malnutrition, and secondary infections. Secondary bacterial infection is one of the most common complications and can progress to septic shock.
Cutaneous PVIn rare cases, mucosal involvement is not observed, despite the presence of both Dsg1 and Dsg3 circulating autoantibodies. The term cutaneous PV is used to refer to this presentation. It is believed that the combination of weakly pathogenic anti-Dgs3 IgG autoanti-body, associated with a potent anti-Dsg1 autoantibody, would explain the site of blistering in this form of PV.35
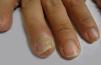
Rare PV manifestationsOther rare clinical manifestations include isolated crusted plaques on the face and scalp, foot ulcers, dyshidrotic eczema, macroglossia, nail dystrophy, paronychia, and subungual hematomas (Figure 6).31,32 Nail involvement usually occurs when the disease is severe and, in most cases, responds partially or completely to systemic therapy.36
Pemphigus vegetans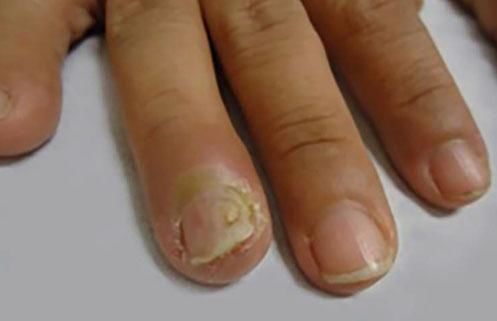
Pemphigus vegetans is a rare clinical variant of PV, accounting for 1%–2% of all cases of pemphigus.37 It is manifested by vegetating plaques with excessive granulation tissue and crusts, especially in the intertriginous areas, face, and scalp. In intertriginous areas, the semi-occlusion, maceration, and mixed infections continuously incite the exudation and formation of granulation tissue.38 Oral involvement is extremely common; occasionally, the tongue may also undergo changes, presenting with cerebriform pattern.31 Verrucous plaques along the vermilion border of the lips or at the angles of the mouth are some presentations of pemphigus vegetans.37 Two clinical subtypes are described: the Neumann type of pemphigus vegetans, which is considered severe and usually begins as PV with vesicles and blisters that rupture to form hypertrophic erosions, evolving into exudative vegetating masses, and the Hallo-peau type of pemphigus vegetans, a milder form than begins with pustules that rupture and evolve into vegetating erosions.38
Neonatal pemphigusNeonatal pemphigus occurs in 30% to 45% of children of PV carriers, by the passage of maternal antibodies to the fetus through the placenta.39 It is manifested by vesicles, blisters, and erosions from the moment of birth, and the involvement of mucous membranes is rare.40 Neonatal pemphigus is transient and tends to disappear spontaneously within three weeks, as it results from the transfer of antibodies that are progressively eliminated.
Differential DiagnosisEstablishing the diagnosis in patients who present only oral lesions is more difficult than in those with the mucocutaneous condition. In PV, oral erosions may mimic several diseases, such as aphthous stomatitis, acute herpetic stomatitis, erythema multiforme or Stevens-Johnson syndrome, lichen planus, systemic lupus erythematosus, paraneoplastic pemphigus, and mucous membrane pemphigoid.28,31,32 The blisters do not last long in the mouth and the biopsy of erosions often do not allow diagnosis. Direct immunofluo-rescence is the most accurate method for the diagnosis of mucosal pemphigus.28
For cutaneous lesions of PV, the differential diagnosis includes other forms of pemphigus, bullous pemphigoid, linear IgA bullous dermatosis, bullous erythema multiforme, and dermatitis herpetiformis.
Intertrigimous lesions of pemphigus vegetans should be differentiated from chronic infections and Hailey-Hailey disease. Vegetating plaques simulating pemphigus vegetans can also be seen in IgA pemphigus and in paraneoplastic pemphigus. The histologic differential diagnosis includes Hailey-Hailey disease, Darier's disease and Grover's disease, or transient acantholytic dermatosis.38
Laboratory DiagnosisFor the laboratory diagnosis of PV, Tzanck smear, histopathological examination, direct immunofluorescence examination, or even immunohistochemical examination may be used.
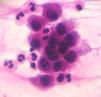
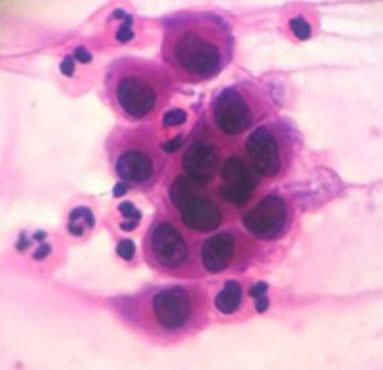
Cytological examinationCytological examination (Tzanck smear) is useful for the rapid demonstration of acantholytic keratinocytes of the spinous layer (abundant eosinophilic cytoplasm and rounded central nucleus), stained preferably by hematoxylin and eosin (Figure 7).31
Histopathological examination helps to identify the level of blister cleavage in order to diagnose pemphigus, and to differentiate with other subepidermal bullous lesions, as acantholytic keratinocytes can be observed in several vesiculobullous diseases (Hailey-Hailey-like Grover's disease and Hailey-Hailey disease, among others).41 For the biopsy, it is recommended to choose a recent blister (less than 24 hours of appearance) that fits inside a 4 mm punch or a small fusiform excision, because PV blisters usually rupture easily. If this is not possible, a perilesional area should be biopsied, so that the blister roof is attached to the adjacent skin and does not detach during histological processing. The biopsy should be fixed in 10% formalin (buffered formalin is better for surface antigen preservation). Histopathological examination indicates the level of epidermal cleavage (suprabasal or intramalpighian). Its contents can often be lost during histological processing or inflammatory cells, with predominance of neutrophils and eventually a few eosinophils can be observed (Figure 8). Acantholysis may affect the adnexal epithelium (usually the follicular epithelium), which facilitates the differential diagnosis with Hailey-Hailey disease. In the papillary dermis, an inflammatory infiltrate is observed, with predominance of neutrophils in the perivascular region.
Direct immunofluorescence examination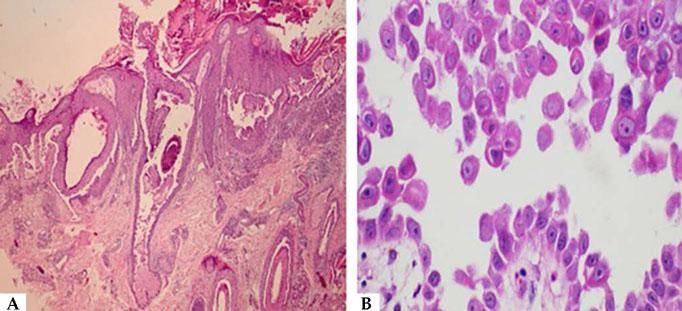
This examination is based on the in vitro antigen-antibody reaction, revealed by ultraviolet-excited fluorochromes (fluorescein isothiocyanate). When tissue deposition of the searched product occurs, the fluorochrome will shine (apple green color).
The identification of IgG and C3 autoantibodies directed against the cell surface of keratinocytes is considered by some authors as a "gold standard" for the differential diagnosis of PV.31,41,42 The most widely used methods for detecting pemphigus autoanti-bodies include direct (DIF) and indirect immunofluorescence (IIF), immunoprecipitation, immunoblotting, and enzyme-linked immunosorbent assay (ELISA).41
In DIF, patients' skin or mucosa may be used to demonstrate IgG and C3 deposits with intercellular distribution (Figure 9). A new biopsy of the perilesional or mucosal skin should be performed and the material should be immediately frozen in liquid nitrogen or placed in a suitable transport medium (Michel's medium).42 It is composed of ammonium sulfate, N-ethyl maleimide, and magnesium sulfate in citrate buffer, which allows specimen preservation for up to two weeks.42 IgG autoantibodies are directed against Dsg3, an autoantigen of higher expression in the lower portions of the epidermis (Figure 9). In case of mucocutaneous lesions, patients may also present Dsg1 antibodies.
Indirect immunofluorescence examination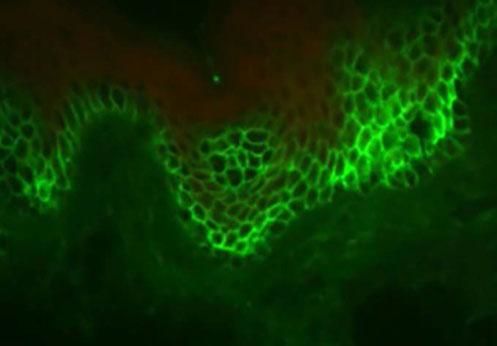
This test assists in the diagnosis of PV and allows the detection of circulating autoantibodies. The normal skin of another individual (originating from the foreskin, breast, or eyelid, which are easy to obtain and present good antigenicity) or a apecimen of monkey esophagus are used as substrate.42 Patient serum is diluted from 1:20 and incubated with the substrate. The reaction is revealed by anti-human (IgG) secondary antibodies produced in animals and conjugated to fluorescein isothiocyanate. The reaction is read under epiluminescence microscopy. In quantitative tests, the resulting titer is the highest at which substrate fluorescence is still detected.43,44 The fluorescence pattern is similar to that of PF. The positivity rate for antiepithelial antibodies of the IgG class ranges from 75% to 100% (Figure 10). A prevalence of IgG4 is observed in active disease.45,46
Immunohistochemical examination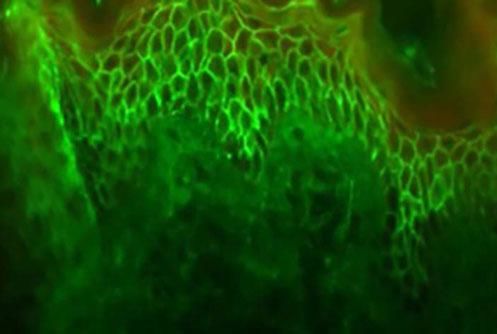
Immunohistochemical examination consists of a combination of immunological and histological methods for the detection of specific antigens in tissues or cells (immunocytochemistry), based on the identification of the antigen-antibody complex.
The most frequently used material for this examination is obtained from histological sections of the paraffin embeded skin biopsy (subjected to the usual technical processing) on silanized slides (containing silane, which helps adherence of the tissue section to the slide, hindering its detachment during the immunohistochemical reaction). The great advantage for the patient is that, if there is already a paraffin embeded skin biopsy and this is representative of the desired site for investigation, this exam can be performed without the need for a new biopsy.47,48 Markers for the detection of intercellular IgG and C3 can be used in PV (Figure 11).
Serological diagnosis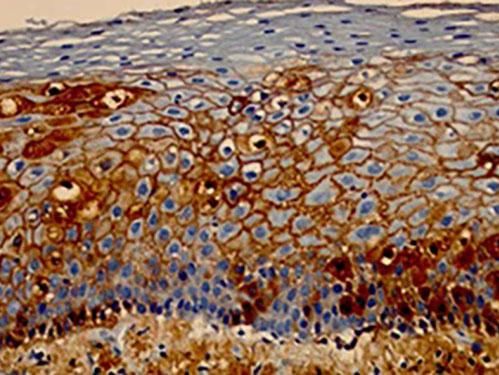
ELISA is a very sensitive and specific method that allows detection of IgG anti-Dsg1 (mucocutaneous PV) and anti-Dsg3 (mucosal PV) autoantibodies in over 90% of patients using recombinant Dsg1 and Dsg3. It is a quantitative method whose result shows a good correlation with clinical severity, and may be useful for patient follow-up.7,49
Immunoblotting and immunoprecipitation are other available serological tests; however, due to their complexity and cost, they are not very useful in clinical practice, being more used in research.
TreatmentThe treatment of autoimmune bullous dermatoses in general and PV in particular is always based on the use of systemic medications (oral or intravenous), as they are severe mucocutaneous mucosal diseases with significant morbidity and mortality rates. Treatment should be initiated as early as possible, aiming to achieve and maintain disease remission. For this, treatment is often quite prolonged, and can last many years (mean: 5 to 10 years). Due to the rarity of PV, few randomized controlled clinical trials have been conducted. However, numerous observational studies, case reports, and case series have been published, supporting the clinical practice of specialists. PV mortality has been very low in the last 50 years; currently, it is caused mainly by the side effects of the medications.50,51
Pre-treatment assessmentClinical assessment: weight, height, blood pressure.
Laboratory tests: blood count; electrolytes; hepatic and renal function; glycemia and glycated hemoglobin; vitamin D; lipids; serology for hepatitis B and C, syphilis and HIV; routine urine test; pregnancy test when appropriate; and chest X-ray and bone densitometry (which should be repeated after six months and annually thereafter).
Ophthalmologic evaluation: initial and annually thereafter.
Systemic treatmentCorticosteroidsSystemic corticosteroids are the basis of PV treatment, as they present potent anti-inflammatory and immunosuppressive action. The introduction of this drug in the 1950s was followed by a reduction in mortality from 75% to 30%.51–53
Oral administrationPrednisone is the most commonly used oral corticosteroid, followed by prednisolone and deflazacort. Although some authors prefer doses of 40 to 60 mg/day (prednisone) for patients with mild PV and 60 to 100 mg/day for more severe conditions, most authors prefer to administer full doses (1 to 2 mg/kg/day oral) for all patients from the beginning, thus avoiding a progressive dose increase. However, the previously used extremely high doses (3 to 4 mg/kg/day) have been shown to be disadvantageous due to frequent and severe side effects. The action of corticosteroids is rapid in PV; improvements are observed within a few days and new lesions cease to appear after two to three weeks. Complete re-epithelization may take up to two months. When the condition is controlled, defined as the interruption of the appearance of new lesions and the total re-e-pithelialization of existing lesions, the corticosteroid dose should be slowly reduced. The reduction should be faster at the beginning and slower at the end; the withdrawal process may take years, and there are no uniform protocols. Some authors recommend that, starting at a given daily dose (usually 40mg/day of prednisone), administration should be made on alternate days, which would minimize side effects. Similarly, there is no consensus on how to increase the dose in case of relapse. Relapses are usually milder than the initial presentation of the disease, requiring equal or lower prednisone doses than those used for initial control.54,55
Pulse therapyCorticosteroids may also be administered in the form of pulse therapy in cases where control with prednisone above 1 mg/kg/day cannot be achieved. Methylprednisolone (1 g/day IV) and dexamethasone (300 mg/day IV) are used, both for three consecutive days. The advantage of pulse therapy is that it allows a faster prednisone dose reduction, minimizing side effects.49,56
Although corticosteroids are quite effective in controlling PV in most patients, they have frequent and potentially serious side effects. The most important are arterial hypertension, diabetes mellitus, cutaneous and systemic infections, gastric ulcer, osteoporosis, femoral head necrosis, glaucoma, and cortisone cataract. The side effects of corticosteroids are partly responsible for the morbidity and mortality of the disease; they also are often responsible for increasing the frequency of consultations, laboratory tests, and hospitalizations. All patients should receive gastric mucosal protectors and vitamin D supplementation.57
In order to minimize these side effects and the morbidity and mortality of PV, and contrary to what was advocated a few decades ago, currently it is recommended that the daily dose of prednisone does not exceed 1.5mg/kg/day, as with higher doses the chance of skin infection and evolution to septicemia (the main cause of death of these patients) increases progressively. Thus it is recommended to use other drugs associated with corticosteroids, which for this reason are called adjuvant or corticosteroid-sparing agents.58
Adjuvant drugsWhen the condition cannot be controlled with corticosteroids alone, or when the patient has clinical contraindications for high dose corticosteroids (e.g., hypertension, diabetes mellitus, glaucoma, or osteoporosis, all of which are relatively frequent in the age group in which PV is most prevalent), other adjuvant or corticosteroid-sparing agents should be associated. Adjuvant drugs have also been used to prevent relapses in previously controlled patients.59
AzathioprineAzathioprine (AZA) is a cytotoxic drug used in most autoimmune diseases. It is an imidazole derivative of mercaptopurine, which antagonizes purine metabolism and inhibits the synthesis of DNA, RNA, and proteins. It may also interfere with cellular metabolism and inhibit mitosis. In addition to this effect on nucleic acid synthesis, AZA also affects the immune system in several other ways. It reversibly reduces the number of monocytes and Langerhans cells; it also interferes with gamma globulin synthesis and T-lymphocyte function, as well as with T helper cell-dependent B cell response and suppressor B cell function.60
The efficacy of AZA as a corticosteroid sparer in autoimmune bullous diseases, particularly in PV, is well documented; it is the oldest and most prescribed immunosuppressive medication for this condition.55,61,62
The AZA dose recommended in PV is 100–200 mg/day (1 to 3 mg/kg/day), orally, divided into two doses. It takes four to six weeks to achieve full therapeutic effect, restricting its use as mono-therapy. In cases of unsatisfactory clinical response, it is recommended to continue the use for three months before replacing it with another adjuvant.54,63
Its main side effects are leukopenia, thrombocytopenia, anemia, pancytopenia, and hepatotoxicity. Prolonged immunosuppression may increase the risk of infections and neoplasms. Individuals with genetic deficiency of the tiopurine methyltransferase enzyme present greater sensitivity to the myelotoxicity of AZA. This drug is contraindicated in pregnant and lactating women.53
Mycophenolate mofetil/sodiumFollowing oral administration, mycophenolate mofetil/sodium (MMF) is absorbed and converted to its active metabolite, mycophenolic acid. This, in turn, selectively inhibits inosine monophosphate dehydrogenase, inhibiting purine synthesis in B and T cells, resulting in an inhibition of proliferation of these cells.64
It has been used as a corticosteroid adjuvant in patients with PV, both as first choice and in patients not responsive to AZA. Some authors prefer MMF to AZA as first-line adjuvant therapy in PV, given the lower hepatotoxicity and the same efficacy. Compared to AZA, MMF would be inferior as a corticosteroid-sparing agent, but more effective in inducing PV control.54,65–67
In PV, the recommended dose is 2–3g/day, divided into two doses. The main side effects are changes in bowel habits, neutropenia, lymphopenia, and myalgia. Therapeutic failure should be considered only after three months of treatment, at a dose of 3 g/day.65,68
RituximabChimeric monoclonal anti-CD 20 antibody (which depletes both normal and pathogenic B lymphocytes) has been used in severe and refractory cases of PV since 2006.69 Following administration of rituximab, a rapid and sustained depletion of circulating and tissue B lymphocytes that persists for at least 6 to 12 months is observed. Recent evidence demonstrates that it also affects T lymphocytes.70 In June 2018, the Food and Drug Administration (United States) approved the use of rituximab for PV treatment. Various prospective and retrospective studies demonstrated its efficacy, which leads to complete and sustained remission in most patients within 3 to 4 months.69,71–74 A recent systematic review, which included 114 studies and 1,085 patients, concluded that rituximab appeared to be an excellent treatment for refractory cases. It should be administered IV, in a slow infusion (four to six hours).75 There are no standardized protocols for the use of rituximab in autoimmune bullous diseases. The literature features studies using both the lymphoma protocol (375 mg/m2, 1 ×/week for four weeks) and the protocol for rheumatoid arthritis (1,000 mg at intervals of two weeks and may be repeated after six months).71,76–78 No differences were observed regarding remission percentage and disease-free time using these two protocols. They can be used alone or in combination with intravenous immunoglobulin, plasmapheresis, and immunoadsorption; the latter two options appear to prolong the response time when compared with rituximab alone. It may also be administered to patients already taking prednisone and immunosuppressive drugs; dose reduction and suspension of the latter should be accelerated, due to the increased risk of infection.75,79–84
Rituximab is generally well tolerated, and serious adverse events are rare. Infusion reactions, which may be reduced with previous administration of analgesics, antihistamines, and corticosteroids, include anaphylaxis, fever, hypotension, chills, headache, nausea, pruritus, and skin rash. Furthermore, neutropenia, hypogammaglobulinemia, and infections, including sepsis, are rarely reported. Some authors and expert groups have already recommended rituximab as a first-line treatment option for PV.54,71,73–75,85–90
CyclophosphamideThis alkylating agent selectively affects B lymphocytes and antibody production. For PV, it can be administered orally (1 to 3 mg/kg/day) or intravenously, sometimes associated with dexamethasone IV, in the form of pulse therapy.91 In this case, dexamethasone is given at a dose of 100mg/day IV for three days, and cyclophosphamide 500mg/day IV is also given on the first day. This pulse therapy is repeated every two to four weeks; between these sessions, the oral dose of cyclophosphamide of 50mg/day and prednisone 1mg/kg/day is maintained. Treatment is considered to have failed after three months of use at a dose of 2mg/kg/day.55,71,92
Its main toxic effects are infertility, predisposition to neoplasias, lymphopenia, and sepsis. Due to its higher toxicity, it should be considered as an adjuvant drug only in cases refractory to AZA and MMF.55,61,93–97
MethotrexateWith an anti-inflammatory action and inhibition of cell proliferation through the inhibition of dihydrofolate reductase, it may be an adjuvant option in the treatment of PV at a dose of 10 to 20mg/week in case of therapeutic failure of other adjuvants. The most frequent side effects are gastrointestinal intolerance, hematological toxicity, and infection.98–101
DapsoneThis drug has anti-inflammatory and anti-TNF action, and can be tried as adjuvant medication in the PV, at a dose of 50 to 200mg/day, orally, with conflicting reports in the literature. Its side effects are usually dose-dependent and reversible.55,102,103
CyclosporineIn rare cases, calcineurin inhibitor with potent immunosu-ppressive action on B and T lymphocytes has been shown to be effective as an adjuvant in the treatment of PV at a dose of 3 to 5mg/kg/day, VO or IV.104 Recently, it has been very little used for PV treatment.
Intravenous human immunoglobulin (IVIG)Derived from a pool of donors, its mode of action in PV is complex, with several mechanisms acting synergistically: it selectively removes pathogenic antibodies; alters the expression and function of Fc receptors; affects the activation, differentiation and effector functions of T and B cells; and interferes with the activation of cytokines and complement. Its advantage is the safety profile, with few side effects (headache, dyspnea, tachycardia, and abdominal discomfort). It is used in PV that fails to respond to other treatments or when there are serious side effects, and has been shown to be effective in some cases at a dose of 0.4g/kg/day for five days, always as an adjunct to corticosteroid therapy once a month. It is a quite expensive medication, and on average three to six cycles are required. It can be used in pregnant women.67,105–107
Anti-TNF drugsTNF-α is one of the cytokines involved in acantholysis. Case reports with the use of infliximab and etanercept have suggested its possible efficacy in PV. However, other studies contradict this possible efficacy.53,108
Plasmapheresis/immunoadsorptionPlasmapheresis was first used in 1978 for the treatment of PV, in order to remove pathogenic autoantibodies from the circulation. However, it was found to trigger a rebound effect, with higher production of these autoantibodies after they were removed from circulation. Therefore, it is recommended that it be associated with corticosteroids and immunosuppressants (e.g., pulse therapy with methylprednisolone and cyclophosphamide) in monthly cycles for up to one year.54,108 IVIG may be used in place of cyclophosphamide to prevent rebound production of autoantibodies. Plasmapheresis is an exceptionally used alternative in the treatment of severe cases of PV that fail to respond to other therapeutic modalities.53 It is available in few hospitals at a very high cost. Its main side effect is septicemia.
Immunoadsorption, introduced in 1984, is a more selective method, which unlike plasmapheresis does not remove other antibodies and plasma components from circulation. Performed in cycles of four consecutive days every four weeks, it has fewer side effects than plasmapheresis.109,110
Systemic antibiotic therapyIt is indicated only in cases with clinical and/or laboratory evidence of secondary infection, never prophylactically. Preferably, the choice of this treatment should be guided by culture and antibiogram of blood and skin samples.
Topical treatmentAlways adjuvant to systemic treatment, topical treatment of PV lesions aims to reduce pain and prevent secondary infection. It is usually performed with corticosteroid and/or antibiotic creams. There have been reports of tacrolimus use, particularly in facial lesions.111 In very extensive cases, antiseptic solutions such as potassium permanganate (1:10,000 or 1:20,000) or chlorhexidine may be used. More potent gel corticosteroids (clobetasol dipropionate) may be used in the oral mucosa. Triamcinolone acetonide (10 mg/mL) may be used in the form of intralesional injection for refractory skin lesions (e.g., pemphigus vegetans).53,54
Future therapiesNew anti-B-cell immunobiological drugs are being investigated in clinical research regarding their efficacy, safety, and cost for patients with PV. These include veltuzumab (anti-CD20 subcutaneous administration antibody), obinutuzumab, ofatumumab, ocaratuzumab, PRO 121921, anti-BAFF, and anti-BAFF-R.71
Treatment planPV treatment should include two phases, induction and maintenance of remission.55,112–116
Induction of remissionAt this phase, the objective is to control the condition, interrupting the appearance of new bullous lesions and promoting re-epithelialization of the existing lesions. Corticosteroids are the most effective and quickest therapeutic option in PV control, being essential at this stage. It may take several weeks to achieve control (on average, three weeks) and dose increasing may be required for this to occur.
Adjuvant medications may be initiated at this stage, but their benefit is limited, as their onset of action is much slower. Thus, isolated use of adjuvant drugs for initial control of the PV is not recommended.
Drug doses should be maintained until the condition is controlled, defined as re-epithelialization of approximately 80% of skin and mucosal lesions, and no new lesions for at least two weeks. Oral mucosal lesions usually present slower resolution than skin lesions. From this moment onwards, the corticosteroid dose can be slowly reduced.
Maintenance of remissionThe drug doses are slowly reduced to minimize the side effects. The ultimate goal is to keep the disease controlled with a prednisone dose of up to 10mg/day. PV is a chronic disease; in one study, 36% of patients received treatment for over 10 years. At this stage, adjuvant medications play a larger role; nonetheless, to date there are no prospective controlled studies that clearly demonstrate the beneficial role of these drugs. For this reason, many authors do not routinely use them in the treatment of PV, unless there are significant contraindications or side effects associated with the use of corticosteroids, or in cases of recurrence when the dose is reduced. Rituximab is an exception; in 2017, the first randomized controlled trial was published, demonstrating the superiority of its combination with prednisolone over prednisolone alone in the PV control after two years (89% vs. 28% complete remission).
Treatment withdrawalComplete remission of the disease is possible, and has been observed in 38%, 50%, and 75% of the cases after three, five, and ten years of diagnosis, respectively. Another study observed that 59% of patients were not being treated three years after diagnosis. However, premature withdrawal should be avoided, being rarely possible before one year.
Evolution and PrognosisBefore the advent of corticosteroids and immunosuppressants, the two-year mortality rate of PV was 50%. Currently, the mortality rate is approximately 10%. The main cause of death in PV patients is septicemia. Patients often evolve as major burn victims, with loss of the skin-mucosal barrier, favoring hydroelectrolytic and metabolic infections and disorders. Oral lesions are usually more resistant to treatment; they may persist for years, significantly impairing the patients' quality of life. It is often possible to achieve total disease control, which allows withdrawing the medication, but patients should be kept under observation, since relapses are frequent.63,117–119
As PV is a rare disease, it is very difficult to compare the efficacy of PV control and relapse prevention, as well as side effects and morbidity and mortality, in the published studies with the different adjuvant drugs. This is due to differences in study design, populations studied, and the doses and combinations used, and mainly due to the lack of randomized controlled clinical trials. Recent systematic reviews and meta-analyses are conclusive regarding the importance of systemic corticosteroids (prednisone or prednisolone) as the basis of PV treatment, but inconclusive as to the best initial corticosteroid dose and the best adjuvant drug.59,67,93,120 Some studies have compared different doses of prednisolone, IV corticosteroids vs. placebo, AZA vs. MMF, and use of other adjuvant therapies such as methotrexate, cyclosporine, cyclophosphamide, and IVIG in high doses.67,114 Despite the lack of a definitive support in the literature, most authors consider the combination of systemic corticosteroids (prednisolone 1-1.5mg/kg/day) with corticosteroid-sparing adjuvant drugs (mainly AZA and MMF) as the first-line standard therapy for PV.91 Some authors and expert groups have already recommended rituximab as a first-line treatment option for PV 54,71,73–75,85,90
Questions- 1.
The following conditions may be associated with pemphigus vulgaris, except:
- a)
Cardiovascular diseases, systemic lupus erythematosus, multiple myeloma, and depression
- b)
Rheumatoid arthritis, myasthenia gravis, non-Hodgkin's lymphoma, and type 1 diabetes mellitus
- c)
Esophageal neoplasia, laryngeal neoplasia, chronic leukemias, and polycythemia vera
- d)
Hydradenitis, smoking, insomnia, and Parkinson's disease
- a)
- 2.
Mark the correct alternative regarding the "desmoglein compensation theory":
- a)
The absence of mucosal lesions in pemphigus foliaceus is justified by the high concentration of desmoglein 1 (Dsg1) throughout the mucosal epithelium
- b)
In mucosal pemphigus vulgaris, the absence of skin lesions is due to the exclusive presence of Dsg1 in the cutaneous epithelium
- c)
In mucocutaneous pemphigus vulgaris, there is a predominance of mucosal lesions, since the only antigen involved is Dsg3
- d)
In the skin, Dsg1 is expressed more intensely in the superficial layers of the epidermis, while Dsg3 is concentrated in the lower layers
- a)
- 3.
Check the correct alternative regarding pemphigus vulgaris:
- a)
The first clinical manifestations are observed in the skin;
- b)
Intact blisters are often found in the oral mucosa
- c)
Paronychia and nail dystrophy may be observed in pemphigus vulgaris
- d)
Oral involvement is rare in pemphigus vegetans
- a)
- 4.
Regarding pemphigus vulgaris, mark the incorrect alternative:
- a)
It is the most severe form of pemphigus
- b)
In neonatal pemphigus, skin lesions are transient
- c)
Lichen planus, systemic lupus erythematosus, and erythema multiforme are differential diagnoses of mucosal pemphigus vulgaris
- d)
Oral involvement is characterized by painless erosions mainly on the palatal and buccal mucosas, lips, and gingiva
- a)
- 5.
The most sensitive and specific test to confirm the diagnosis of pemphigus vulgaris is:
- a)
Tzanck smear
- b)
Biopsy and histopathological examination
- c)
Immunohistochemical examination
- d)
Direct immunofluorescence
- a)
- 6.
Regarding the laboratory diagnosis of pemphigus vulgaris, the following statement is correct:
- a)
It presents mainly anti-desmoglein 1 autoantibodies
- b)
The acantholytic blisters are subcorneal
- c)
It may present IgG4 autoantibodies in active disease
- d)
It presents IgG and C3 autoantibodies in the basal membrane zone
- a)
- 7.
Regarding the use of corticosteroids in the treatment of PV, check the correct alternative:
- a)
They are indicated only for mucocutaneous cases, but not in exclusively mucosal forms
- b)
The dose should be promptly increased until complete control of the condition is achieved, and then reduced slowly
- c)
They can be used in doses of up to 4 mg/kg/day, until control of the condition is achieved
- d)
They are the basis for the treatment of PV, and the most used drug is deflazacort
- a)
- 8.
Regarding adjuvant medications in the treatment of PV, check the incorrect alternative:
- a)
Azathioprine should be administered at a dose of 1 to 3 mg/kg/day and may be hepatotoxic
- b)
The dose of mycophenolate mofetil is 2 to 3 g/day, and it usually causes gastrointestinal side effects
- c)
Cyclosporine can be used orally and in the form of pulse therapy
- d)
Methotrexate may be used at a dose of 10 to 20 mg/week
- a)
- 9.
Check the correct alternative regarding the use of rituximab in the treatment of PV:
- a)
It is an anti-TNF-α drug, contraindicated in patients with history of tuberculosis
- b)
The most used scheme is the lymphoma protocol, which uses 1,000 mg/m2, once a week for four to eight weeks
- c)
It may be administered in combination with corticosteroids and immunosuppressants
- d)
It should always be used in combination with intravenous immunoglobulin to avoid a rebound effect
- a)
- 10.
Mark the incorrect alternative regarding the evolution of the PV:
- a)
The mortality rate fell from 50% to 10% with the use of systemic corticosteroids
- b)
The main cause of death is infection; therefore, it is always necessary to introduce prophylactic antibiotic therapy together with immunosuppressants
- c)
Mucosal lesions usually appear before cutaneous lesions, and respond more slowly to treatment
- d)
It is rarely possible to stop treatment in less than one year
- a)
Answers
Bullous pemphigoid. An Bras Dermatol. 2019;94(2):133–46.
1. B
2. D
3. C
4. A
5. A
6. D
7. B
8. C
9. C
10. D
